





Abstract
There is a great deal of interest in developing less invasive markers for monitoring airway inflammation and the effect of possible novel anti-inflammatory therapies that may take time to impact on disease pathology. Exhaled nitric oxide (eNO) has been shown to be a reproducible, noninvasive indicator of the inflammatory status of the airway in the clinic.
The aim of the present study was to determine the usefulness of measuring eNO as a marker of the anti-inflammatory impact of glucocorticoid and an inhibitor of κB kinase-2 (IKK-2) inhibitor 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1), in a pre-clinical model of airway inflammation. Rats were given vehicle, budesonide or TPCA-1 prior to exposure to lipopolysaccharide, previously shown to induce an increase in eNO and airway neutrophilia/eosinophilia.
Comparison of the effect of the two compounds on inflammatory components demonstrated a significant correlation between the impact on eNO and inflammatory cell burden in the airway.
The current study demonstrates the usefulness of profiling potential disease-modifying therapies on exhaled nitric oxide levels and the way in which an effect on this noninvasive biomarker relates to effects on pathological parameters such as lung cellularity. Information from studies such as the current one would suggest that the measurement of exhaled nitric oxide has potential for monitoring inflammatory status in lung tissue.
Much effort in the pharmaceutical industry is geared towards developing therapies that inhibit inflammation in respiratory diseases such as asthma and chronic obstructive pulmonary disease (COPD), in the belief that this will impact on disease pathology. One issue concerning potential novel anti-inflammatory therapies is that prolonged treatment may be needed to influence disease pathology. Clinical trials (especially when trialling compounds for COPD) are often lengthy studies that require large numbers of patients and are consequently very costly. For this reason it is important to be as confident as possible that the potential new therapy is going to impact on the desired pathology in early proof-of-concept studies before embarking on large-scale clinical trials. This could be achieved by identifying and using disease-relevant biomarkers as early indicators of compound efficacy before embarking on larger-scale clinical trials that document compound effects on functional end-points (e.g. forced expiratory volume in one second, symptom scoring). Another issue arising from new therapies is their potential to promote undesirable side-effects. However, if drug effects on inflammation could be routinely monitored in a simple, noninvasive fashion, then it may be possible to titrate the amount administered to a just-effective dose, which may reduce the risk of any unwanted side-effects. Regular invasive assessment of lung inflammation (e.g. by measurement of biomarkers in bronchiolar lavage or lung biopsy samples) is uncomfortable for the patient and in certain situations, such as severe asthma and COPD, can be dangerous, exposing patients to an increased risk of infection. Furthermore, the small samples of lung tissue removed during biopsy procedures may not be representative of the disease pathology observed in the whole lung. Consequently, there is a great deal of interest in developing less invasive markers for monitoring airway inflammation.
Increased levels of exhaled nitric oxide (eNO) have been measured in patients with respiratory diseases such as asthma and COPD 1–3. Therefore, it is believed that eNO could be used as a noninvasive biomarker of respiratory inflammation in the clinic, although one caveat is that in COPD patients the use of eNO is perhaps not straightforward, since the measurements are affected by smoking status 4. The noninvasive nature of this technique means it is amenable to serial measurements, making it a useful and reproducible guide to determining the effectiveness of disease-modifying therapies 5. In asthmatic patients, levels of eNO have been shown to correlate with sputum eosinophilia, bronchoalveolar lavage (BAL) fluid eosinophilia, blood eosinophilia, airway hyperreactivity and peak-flow variability 6–12. In addition, the impact of glucocorticoid therapy on another aspect of the asthma phenotype, airway hyperreactivity, has been shown to be associated with a reduction in eNO levels in asthmatic patients 13. Although other noninvasive biomarkers, such as measurement of mediators in induced sputum and exhaled breath condensate, have been used in academic studies, they are still in their infancy with much more validation work needed before they can be recommended as an end-point in clinical trials or adopted as a disease severity indicator in clinical practice. Recommendations for optimising these methodologies have been the subject of several task forces led by key opinion-leaders 14, 15.
When considering using eNO as a marker of lung inflammation in clinical trials employing possible novel disease-modifying therapies, it is important to determine whether any impact on eNO levels relates to an effect on lung inflammation. The aim of the present study was to profile a clinically relevant glucocorticoid and another anti-inflammatory compound, 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1), in a pre-clinical in vivo model of airway inflammation to determine the utility of eNO as a noninvasive biomarker of lung cellular burden. The pre-clinical model selected was the aerosolised lipopolysaccharide (LPS)-driven rat model, which the current group of authors has previously shown to exhibit increases in eNO and increased cellular burden, and which is sensitive to glucocorticoid therapy 16. The aim of an earlier study 16 was to determine the time and dose–response relationship between aerosolised LPS and eNO and secondly, to determine the role of nitric oxide (NO) in this model using pharmacological tools. This was achieved by investigating the effect of NO synthase (NOS) inhibitors on eNO, neutrophilia and inflammatory biomarkers. Dexamethasone was used as a positive control at a supramaximal concentration. The question addressed in the present manuscript was whether there was a correlation between the impact on cellular inflammation and the eNO measurement using two different classes of anti-inflammatory compound.
TPCA-1 is a potent and selective inhibitor of inhibitor of κB (IκB) kinase-2 (IKK-2) 17. IKK-2 plays a key role in the removal of IκB from nuclear factor (NF)-κB, a transcription factor known to regulate the expression of many inflammatory genes, allowing NF-κB to enter the nucleus and initiate gene transcription. The current authors previously demonstrated the anti-inflammatory properties of TPCA-1 in an antigen-driven rat model 18 and so adopted a similar dosing regimen in the present study.
In the present study, after NOS staining in structural and inflammatory cells after exposure to LPS was demonstrated, the effect of the two anti-inflammatory compounds on eNO, cellular burden in the BAL fluid and lung tissue and a range of cytokines believed to play a role in the inflammatory response in this model (tumour necrosis factor (TNF)-α, interleukin (IL)-1β, cytokine-involved neutrophil chemoattractant (CINC)-1, CINC-2 and CINC-3) 19, 20, was determined in the same rat strain. In this way, the manner in which the impact on eNO related to the anti-inflammatory effect of the compounds of interest could be elucidated.
MATERIALS AND METHODS
Materials
Sodium pentobarbitone (200 mg·kg-1) was obtained from Rhône Mérieux (Harlow, UK) RPMI 1640 medium, buffers and gels for zymography and foetal bovine serum were obtained from Invitrogen (Paisley, UK). Roche Diagnostics (Lewes, UK) supplied DNase and collagenase. LPS, budesonide and Wright's Giemsa stain were obtained from Sigma (Poole, UK). TPCA-1, a potent and selective IKK-2 inhibitor 17, was a gift from GlaxoSmithKline (Stevenage, UK). All ELISA Duoset® kits were from R&D Systems (Abingdon, UK). Endotoxin-free saline was bought from Fresenius Kabi (Warrington, UK). Male Wistar rats (150–180 g) were purchased from Harlan-Olac (Bicester, UK) and kept for ≥5 days before initiating experiments. Food and water were supplied ad libitum. UK Home Office guidelines for animal welfare, based on the Animals (Scientific Procedures) Act 1986, were strictly observed. There were eight animals per experiment.
Effect of LPS exposure on NOS IHC staining in the airways
Immunohistochemical staining for the NOS isoforms was performed on histologically prepared lung tissue from rats 6 h after exposure to saline or LPS. Briefly, after insufflating the lungs with 10% formalin at a pressure of 2.6 kPa, histological lung sections were prepared. The section slides were put in citrate buffer (10 mM, pH 6.0, 2×10 min) in a microwave oven, to expose the epitopes, and then incubated with 3% hydrogen peroxide in methanol (30 min at room temperature) to block endogenous peroxidase. Nonspecific binding of the secondary antibody was reduced by incubating the slides in goat serum (1:6 in Tris-buffered saline containing 4 drops·mL-1 avidin D block). The sections were then incubated overnight at 4°C with rabbit polyclonal primary antibodies directed against rat NOSII (1:2,000 dilution), NOSIII (1:800 dilution), NOSI (1:800 dilution) or rabbit control (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, the sections were exposed to anti-rabbit biotinylated secondary antibody (1:200; Dako, Ely, UK). Following a further wash, the sections were incubated with diaminobenzidine chromogen (Vector Laboratories, Peterborough, UK) for 10 min. The sections were then counter-stained with haematoxylin to help determine cell types.
Effect of budesonide on LPS-induced eNO production and airway inflammation in the Wistar rat
Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (1 mg·mL-1 for 30 min) in a perspex box (600×240×350 mm). The exposure appeared relatively uniform given the reproducibility between animals of the responses obtained 19. However, the absolute LPS dose delivered to the lungs of each animal was not calculated. Vehicle (0.5% methylcellulose and 0.2% Tween 80 in distilled water, 2 mL·kg-1), or budesonide (0.1, 0.3, 1 or 3 mg·kg-1) was administered orally 24 and 1 h pre- and 2 h post-challenge (dosing levels and protocol previously shown to be effective) 18. eNO production was assessed 4 h post-challenge, as outlined below. Cellular inflammation and mediator protein levels (IL-1β, TNF-α, CINC-1, CINC-2 and CINC-3) were determined 6 h post-challenge, as outlined below.
Effect of TPCA-1 on LPS-induced eNO production and airway inflammation in the Wistar rat
Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (1 mg·mL-1 for 30 min) as above. Vehicle (dimethyl sulfoxide (2%), Cremophor EL (10%) and ethanol (5%) in water, 5 mL·kg-1), or TPCA-1 (3, 10, 30 or 60 mg·kg-1) was administered orally 1 h pre- and 2 h post-challenge (dosing levels and protocol previously shown to be effective) 16. eNO production was assessed 4 h post-challenge, as outlined below. Cellular inflammation and mediator protein levels (IL-1β, TNF-α, CINC-1, CINC-2 and CINC-3) were determined 6 h post-challenge, as outlined below.
Measurement of eNO production from Wistar rats following LPS challenge
eNO was measured using the Sievers NO analyser (Analytix, Peterlee, UK). Animals were placed in sealed 3-L Perspex boxes, which had a scrubber attached to the bottom port of the chamber. The top valve of the box was connected by a tube to a pump, which when turned on pulled NO-free air through the scrubber into the box. NO was purged from the box for 10 min, before the pump was turned off and the valve closed, allowing eNO to build up for 20 min. At the end of this time, an eNO measurement was taken using a sampling tube, which was connected to the analyser and could be attached to a port on the front of the box. eNO production was expressed as parts per billion (ppb).
Quantification of airway inflammation
Six hours after saline or LPS challenge animals were euthanised with sodium pentobarbitone (200 mg·kg-1 i.p.) and the trachea cannulated. BAL cells were recovered from the airway lumen by flushing the airways with 10 mL·kg-1 RPMI 1640 medium, delivered through the tracheal cannula and removed after a 30-s interval. This procedure was repeated and samples were pooled for each animal.
The inflammatory cells were extracted from the lung tissue by collagenase digest, as described previously 16. Total white cell numbers in the BAL and lung tissue samples were determined on the Sysmex F820 haematology analyser (Sysmex, Milton Keynes, UK). Cytospins of these samples were prepared by centrifugation of 100 μL aliquots in a cytospin (Shandon, Runcorn, UK) at 700 rpm for 5 min, low acceleration at room temperature. Slides were fixed and stained on a Hema-tek 2000 (Ames Co., Elkhart, IN, USA) with modified Wright’s Giemsa stain. Three-part differential counts on 200 cells per slide were performed following standard morphological criteria and the percentage of eosinophils, lymphomononuclear cells and neutrophils were determined.
Mediator level determination in the BAL supernatant and lung tissue following LPS challenge
For the determination of cytokine protein levels in the lung tissue, ∼200 mg of lung tissue was homogenised in PBS using an Ultra Turrax T25 homogeniser (IKA Werke, Staufen, Germany). The samples were then spun in a benchtop microcentrifuge at 13,000 rpm for 10 min (MSE Micro Centaur; Jencons (Scientific) Ltd, Leighton Buzzard, UK). The resulting supernatants were used for cytokine protein quantification. The level of NOSII gene expression in the lung tissue was determined using real-time TaqMan (Applied Biosystems, Warrington, UK) PCR as described previously 16.
IL-1β, TNFα, CINC-1, CINC-2 and CINC-3 protein levels were determined by ELISA using rat Duosets® (R&D Systems) according to the manufacturer's instructions. Cytokine levels in lung tissue were further corrected for total protein content, measured using the Bradford assay.
Analysis
Values are expressed as mean±sem of independent observations. Impacts on LPS exposure were compared using the T-test with a Mann–Whitney U-post test for unpaired data. Statistical analyses of multiple comparisons were made using the Kruskal–Wallis test, followed by a Dunn's post test. All treatments were compared with relevant vehicle control groups. A p-value <0.05 was regarded as significant.
To calculate the dose of compound evoking 50% of maximal inhibition (ED50) values, the raw data was converted into a percentage inhibition; dose–response curves were constructed and analysed by least-squares nonlinear iterative regression with the PRISM curve fitting program (Graphpad Software, San Diego, CA, USA). ED50 values were subsequently interpolated from curves of best fit. Saline challenge levels were subtracted from all other LPS challenge treatment groups and treatment effect was expressed as percentage of vehicle-treated LPS challenge control and (in the absence of 100% inhibition) the dose necessary to inhibit 50% of the LPS response was calculated.
RESULTS
Effect of LPS exposure on NOS immunohistochemical staining in the airways
Staining for the three NOS isoforms demonstrated a marked increase, predominantly in NOSII levels, after exposure to LPS (fig. 1⇓). The observed profile of increased protein levels of NOSII, compared with NOSIII and NOSI, is very similar to that measured at the gene expression level for these three isoforms previously published by the current group in the same model 16. There appeared to be an increased thickness of the epithelial layer, which was associated with staining for NOSII following the LPS challenge. NOSII (and to a lesser extent NOSI) expression was also evident in the inflammatory cells (neutrophils, macrophages, eosinophils) that were recruited to the lung tissue following LPS challenge (fig. 1⇓). Interestingly, the presence of NOSI and NOSII staining in neutrophils infiltrating into the lung following LPS exposure is consistent with other studies in the literature demonstrating the presence of these NOS isoforms in this cell type 21.

Effect of exposure to lipopolysaccharide (LPS) on nitric oxide synthase (NOS) isoform expression in the airways. Male Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (for 30 min). Lungs were fixed, removed, processed and stained for NOS isoform expression 6 h post-challenge. a, c, e) Examples of staining for NOSI (nasal NOS), NOSII (Inclucible NOS) and NOSIII (eNOS), respectively in saline-challenged rats. b, d, f) Examples of staining for NOSI, NOSII and NOSIII, respectively, in LPS-challenged rats.
Effect of budesonide on LPS-induced eNO production and airway inflammation in the Wistar rat
LPS challenge, compared with saline, caused a significant increase in total white cells in BAL fluid and lung tissue (figs 2⇓ and 3⇓). Differential counts revealed that the increase in total cell number was due to increased recruitment of neutrophils and, to a lesser extent, eosinophils; there was no effect on mononuclear cell number (figs 2⇓ and 3⇓). This increase in cellular burden in the lung was accompanied by a significant increase in eNO (fig. 4⇓) and cytokine levels (TNF-α, CINC-2/3 in the BAL fluid and IL-1β and CINC-1/2/3 in the lung tissue; tables 1⇓ and 2⇓).

Effect of budesonide on lipopolysaccharide (LPS)-induced airway inflammation, as measured using bronchoalveolar lavage fluid. Male Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (1 mg·mL-1 for 30 min). Vehicle (0.5% methylcellulose and 0.2% Tween 80 in distilled water, 2 mL·kg-1; □) or budesonide (0.1, 0.3, 1 or 3 mg·kg-1; ░) was administered orally 24 and 1 h pre- and 2 h post-challenge. Cellular inflammation was determined 6 h post-challenge. a) Total white cell count, b) neutrophil number and c) eosinophil number were determined. Data are presented as mean±sem (n = 8). #: p<0.05 versus saline-challenged vehicle control; *: p<0.05 versus LPS-challenged vehicle control.

Effect of budesonide on lipopolysaccharide (LPS)-induced airway inflammation, as measured using lung tissue. Male Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (1 mg·mL-1 for 30 min). Vehicle (0.5% methylcellulose and 0.2% Tween 80 in distilled water, 2 mL·kg-1; □) or budesonide (0.1, 0.3, 1 or 3 mg·kg-1; ░) was administered orally 24 and 1 h pre- and 2 h post-challenge. Cellular inflammation was determined 6 h post-challenge. a) Total white cell count, b) neutrophil number and c) eosinophil number were determined. Data are presented as mean±sem (n = 8). #: p<0.05 versus saline-challenged vehicle control; *: p<0.05 versus LPS-challenged vehicle control.

Effect of budesonide on lipopolysaccharide (LPS)-induced increase in exhaled nitric oxide (eNO). Male Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (1 mg·mL-1 for 30 min). Vehicle (0.5% methylcellulose and 0.2% Tween 80 in distilled water, 2 mL·kg-1; □) or budesonide (0.1, 0.3, 1 or 3 mg·kg-1; ░) was administered orally 24 and 1 h pre- and 2 h post-challenge. eNO production was assessed 4 h post-challenge. Data are presented as mean±sem (n = 8). #: p<0.05 versus saline-challenged vehicle control; *: p<0.05 versus LPS-challenged vehicle control.
Effect of budesonide on inflammatory mediator release in the bronchoalveolar lavage(BAL) fluid and lung tissue
Effect of 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) on inflammatory mediator release in the bronchoalveolar lavage (BAL) fluid and lung tissue
Treatment with budesonide inhibited LPS-induced cellular recruitment and eNO levels (figs 2⇑–⇑4⇑). The ED50 values for inhibition of total white cells in the BAL fluid and lung tissue were calculated to be 1 and 2 mg·kg-1, respectively, with maximal inhibitions, using this protocol, of 75 and 55%, respectively. The ED50 and maximal inhibition values, using this protocol, for the impact of budesonide treatment on eNO levels were 2 mg·kg-1 and 59%, respectively. This inhibition of eNO was paralleled by an inhibition of NOSII gene expression. LPS challenge alone caused a 42±5-fold increase in expression, which was reduced by 32, 63 or 88% by 0.3, 1 or 3 mg·kg-1 budesonide, respectively. Assessment of the impact of budesonide treatment on cytokine levels showed a reduction in BAL fluid TNF-α and lung tissue IL-1β at the same doses and with a similar magnitude to the BAL fluid cellular recruitment (table 1⇑). One caveat of making associations of this sort is that some may have arisen by chance. Interestingly, budesonide treatment had little impact on the levels of CINC-1/2/3 (tables 1⇑ and 2⇑).
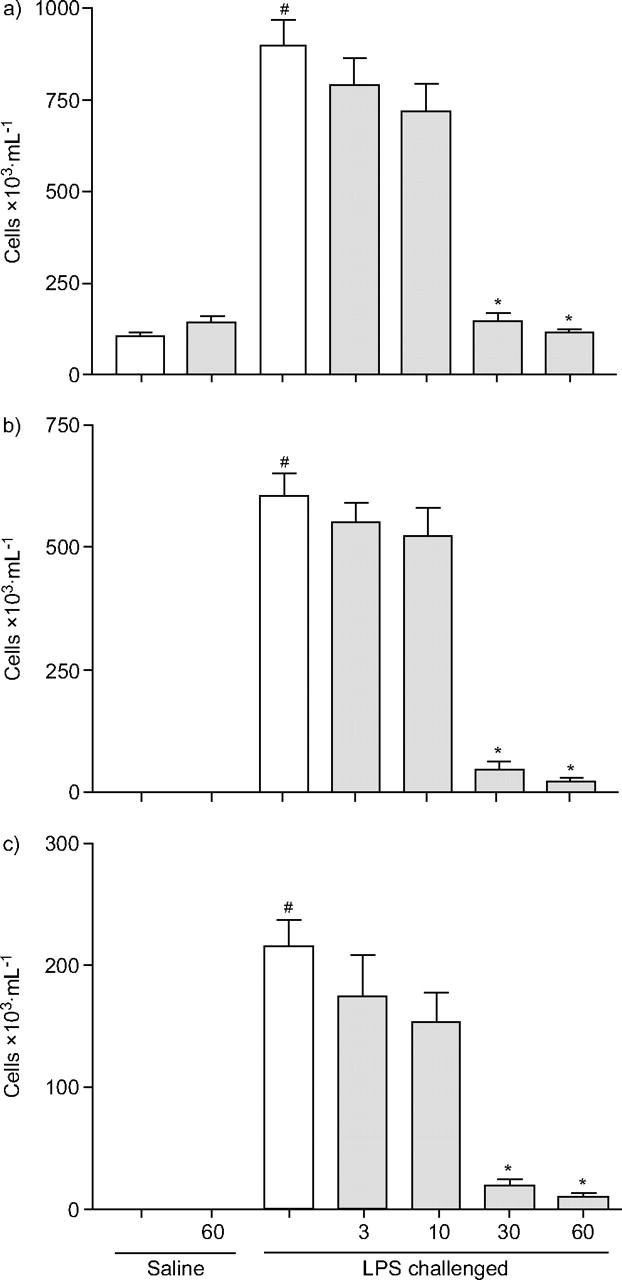
Effect of TPCA-1 on LPS-induced eNO production and airway inflammation in the Wistar rat
LPS challenge, compared with saline, caused a significant increase in total white cells in the BAL fluid and lung tissue (figs 5⇓ and 6⇓). Differential counts revealed that the increase in total cell number was due to increased recruitment of neutrophils and to a lesser extent eosinophils. There was no effect on mononuclear cell number (mononuclear cell data not shown). This increase in cellular burden in the lung was accompanied by a significant increase in eNO (fig. 7⇓) and cytokine levels (TNF-α and CINC-2/3 in the BAL fluid, and IL-1β and CINC-1/2/3 in the lung tissue; table 1⇑).

Effect of 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) on lipopolysaccharide (LPS)-induced airway inflammation, as measured using bronchoalveolar lavage fluid. Male Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (1 mg·mL-1 for 30 min). Vehicle (dimethyl sulfoxide (2%), Cremophor EL (10%) and ethanol (5%) in water, 5 mL·kg-1; □) or TPCA-1 (3, 10, 30 or 60 mg·kg-1; ░) was administered orally 1 h pre- and 2 h post-challenge. Cellular inflammation was determined 6 h post-challenge. a) Total white cell count, b) neutrophil number and c) eosinophil number were determined. Data are presented as mean±sem (n = 8). #: p<0.05 versus saline-challenged vehicle control; *: p<0.05 versus LPS-challenged vehicle control.

Effect of 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) on lipopolysaccharide (LPS)-induced airway inflammation, as measured using lung tissue. Male Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (1 mg·mL-1 for 30 min). Vehicle (dimethyl sulfoxide (2%), Cremophor EL (10%) and ethanol (5%) in water, 5 mL·kg-1; □) or TPCA-1 (3, 10, 30 or 60 mg·kg-1; ░) was administered orally 1 h pre- and 2 h post-challenge. Cellular inflammation was determined 6 h post-challenge. a) Total white cell count, b) neutrophil number and c) eosinophil number were determined. Data are presented as mean±sem (n = 8). #: p<0.05 versus saline-challenged vehicle control; *: p<0.05 versus LPS-challenged vehicle control.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) on lipopolysacharide (LPS)-induced increase in exhaled nitric oxide (eNO). Male Wistar rats were challenged with an aerosol of endotoxin-free saline or LPS (1 mg·mL-1 for 30 min). Vehicle (dimethyl sulfoxide (2%), Cremophor EL (10%) and ethanol (5%) in water, 5 mL·kg-1; □) or TPCA-1 (3, 10, 30 or 60 mg·kg-1; ░) was administered orally 1 h pre- and 2 h post-challenge. eNO production was assessed 4 h post-challenge. Data are presented as mean±sem (n = 8). #: p<0.05 versus saline-challenged vehicle control; *: p<0.05 versus LPS-challenged vehicle control.
Treatment with TPCA-1 inhibited LPS-induced cellular recruitment and eNO levels (figs 5⇑–⇑7⇑). The ED50 values for inhibition of total white cells in the BAL fluid and lung tissue were calculated to be 12 and 16 mg·kg-1, respectively, with maximal inhibitions, using this protocol, of 99 and 100%, respectively. The ED50 and maximal inhibition values, using this protocol, for the impact of TPCA-1 treatment on eNO levels were 10 mg·kg-1 and 89%, respectively. The inhibitory effect of this compound on BAL fluid cells was described by a steep dose–response curve but the reason for this is unknown. This inhibition of eNO was similar to the inhibition of NOSII gene expression. LPS challenge alone caused a 47±11-fold increase in expression, which was reduced by 54, 83 or 99% by 10, 30 or 60 mg·kg-1 TPCA-1, respectively. Assessment of the impact of TPCA-1 treatment on cytokine levels showed a reduction in BAL fluid TNF-α and lung tissue IL-1β at the same doses and with a similar magnitude to the BAL fluid cellular recruitment (table 2⇑). Interestingly, as with budesonide, TPCA-1 treatment had no inhibitory effects on the levels of CINC-1/2/3 (table 2⇑).
Comparison of the effect of both compounds on the various inflammatory components demonstrated parallels between the compounds’ impact on eNO and inflammatory cell burden in the airway (% inhibition of eNO versus % inhibition of total cellular infiltration into BAL fluid and lung tissue). A significant correlation was determined using a Spearman rank correlation for nonparametric data (r2 = 0.879; p<0.0001).
DISCUSSION
The results of the present study suggest an association between the effect of budesonide and the IKK-2 inhibitor TPCA-1 on eNO levels and inflammatory cellular burden in the lung in this pre-clinical model. Indeed, this is reflected by the positive correlations observed between the effect of both compounds on eNO and their effect on inflammatory cells in the airway lumen or lung tissue. This seems particularly apparent for both compounds when comparing the percentage inhibition of eNO and lung tissue cellular burden (table 3⇓), although the relative proportion of tissue versus luminal cells could depend on whether inflammation is assessed during the early recruitment phase (as in the present study) or during late-stage/chronic inflammatory conditions. However, this is an interesting observation, if it translates to the situation in humans, as it has been suggested that inflammation in the lung tissue, which is difficult to assess compared with the cellular inflammation found in the airway lumen, may be more important in disease pathogenesis. Whether associations such as this will translate into other pre-clinical disease-relevant models, or whether the observations can be extrapolated to the situation in humans, is not known. However, data obtained from studies such as the current one could be used as a guide as to whether to use eNO clinically as a noninvasive marker of airway inflammation. Interestingly, when cellular infiltration was assessed in the current LPS model, an increase in eosinophils was noted in the BAL and lung tissue compartments. Interestingly, eosinophilia has been reported previously both in animal models and in humans exposed to LPS 22, 23.
Effect of budesonide and 2-[(aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide (TPCA-1) on inflammatory mediator release in the bronchoalveolar lavage (BAL) fluid and lung tissue
The use of budesonide and TPCA-1 as tools to explore the utility of eNO as a marker of airway inflammation may be complicated because of the direct effect of the compounds on NO production. The present authors have previously shown in this model that the main isoform of NOS responsible for the LPS-induced rise in eNO is NOSII 16, a result which appears to be confirmed by the NOS isoform staining presented in the current study. In the same study 16 and using NOS inhibitors, the current authors demonstrated that NO plays a small role in cellular recruitment. Therefore, the effect of the compounds in the present study is unlikely to be solely due to reducing NO production. Indeed, it is likely that compounds such as these inhibit a multitude of pro-inflammatory mediators at the same dose, which may explain why there is a closer parallel between inhibition of eNO and cellular recruitment than between eNO and inhibition of NOSII gene expression. However, it would be interesting to profile other therapies that reduce cellular burden, without impacting directly on NO production, in this system, in order to assess the link between the reduction in cellular inflammation and eNO. Furthermore, the present study highlights the need to test any potential novel therapies in models of this type, as parallels cannot be made with therapies which use different mechanisms of action.
At doses shown to impact on cellular recruitment and TNF-α/IL-1β production, neither compound used in the present study affected CINC-1/2/3 production. As TPCA-1 inhibits NF-κB translocation to the nucleus and glucocorticoids are believed to impact on the actions of pro-inflammatory transcription factors such as NF-κB 24, it seems likely that the NF-κB transcription factor is not involved in CINC production. CINC proteins have homology to growth-related oncogene-α in humans and are inflammatory cell chemoattractants. Therefore, the present results would suggest that the compounds are inhibiting cellular recruitment independently of these chemoattractants, which is possible through their impact on other inflammatory mediators such as TNF-α, IL-1β, adhesion molecules or other chemoattractants.
One caveat to the results of the present study is that it is possible that the sampling technique used for eNO is not specific for NO from the lung. A previous paper by the current authors 16 has detailed discussions as to other possible sources of the NO measured and their relevance. It is possible that the NO measured could be from another source (e.g. intestinal mucosa and flora, bacterial skin flora and nasal passages) and that the drugs are having an effect on NO levels from these sources. A method that allows more lung-specific sampling of NO could address these issues, but this would probably involve invasive techniques and would defeat the aim of obtaining a noninvasive method for assessing airway inflammation. In addition, it appears that the measurement of eNO from nonairway sources is unlikely in this model, given that aerosolised LPS produces a lung-specific inflammatory response with no increase in systemic biomarkers (e.g. TNF-α).
In conclusion, and with all the caveats listed above, the present authors believe that pre-clinical profiling may be valuable in deciding the relevance and usefulness of exhaled nitric oxide as a noninvasive biomarker for monitoring the activity of anti-inflammatory drugs on lung inflammation.
Acknowledgments
The authors would like to thank S. Bottoms and R. McAnulty for the histological processing and staining of the nitric oxide synthase isoforms. They are also very grateful to S. Sham for help with the preparation of this manuscript.
- Received April 7, 2006.
- Accepted August 25, 2006.
- © ERS Journals Ltd
References










