





In this issue of the journal, Prof. Jon Wakefield provides a contribution to the complex topic of screening genetic associations.1 My comments here are intended to clarify some points and outline connections of his discussion to broader problems, describing how methods such as Wakefield's can be appropriate in epidemiology beyond genetic research. I will also comment on a few aspects of his presentation related to technical issues. I will assume (as does Wakefield) that the reader is familiar with the terminology of Bayesian statistics2 as well that of conventional (frequentist) statistics.
I will start with a comment on interpretation of P-values, and then turn to the issue of multiple comparisons.
Proper interpretation of P-values and confidence intervals
As Wakefield mentions, for continuous distributions a valid frequentist P-value is uniformly distributed under the null hypothesis. From this fact, we can deduce that, under the null and assuming no bias, 0.3% of the P-values will be 0.003 or less over repeated sampling. When citing Sellke et al.3 Wakefield might seem to claim that this deduction is incorrect. But the deduction is correct, whether or not we happen to observe P = 0.003, since it refers to repeated sampling under the null, not to the observed data set alone.
To sort out the issues of concern it helps to keep P-values distinct from α- levels, for the two are very different concepts and arise from entirely different schools of frequentist inference. The P-value originated several centuries ago (albeit under other names;4,,5) and only later became the centerpiece of R.A. Fisher's significance testing approach, which entails no mathematical decision rule.6–9 In contrast, the α-level is the maximum allowable Type I error rate for a test; it originated in the 1920s in Neyman and Pearson's decision-theoretic approach to statistical hypothesis testing.7–10
Among valid concerns underlying Wakefield's comment is that we might make claims about a P-value that was picked from multiple ones based on its magnitude (e.g. the smallest P observed), or alter our decision rule upon seeing the P-value (e.g. switch from a 0.05 α-level to a 0.10 α-level if and only if 0.05 < P < 0.10). Such misuse seems invited when someone reports a P-value result by saying the observation is ‘significant at the P-level’, as Fisher sometimes did (albeit he began this practice before Neyman and Pearson's work appeared).4–6
But α-levels and P-values are so closely related mathematically that their confusion seems inevitable, especially because both have been called ‘significance levels’ and ‘critical levels’ by authors who know the distinction perfectly well. In his classic book on statistical testing,10 Erich Lehmann laid out their correct relation within the Neyman–Pearson theory. I think the following points provide an accurate summary of his more symbolic prose:
Imagine we are considering an unlimited sequence of randomized studies (randomized experiments or random-sample surveys) that generate a corresponding sequence of data sets. From each of those we may compute a decision (‘hypothesis test’).
Once we settle on a decision (‘testing’) method appropriate to the study designs in the sequence, each α-level will define a different decision rule for ‘accepting’ or ‘rejecting’ a given test hypothesis. We thus have an infinite number of decision rules, one for each alpha.
All these rules are defined without reference to any particular data set in the sequence.
We can take a given data set and see what each decision rule would yield from that data set.
The P-value from a given data set is the boundary between the α-rules that ‘accept’ and the α-rules that ‘reject’ the test hypothesis when they are fed this data set.
The last point is crucial. In fact, unlike Fisher and many other authors since,1,,3,7–9 Lehmann defines the P-value as this boundary alpha10 (p. 70). Yet, I fear Wakefield 1 and Sellke et al.3 could be read as if they were making the absurd claim that Lehmann's final point is wrong. True, if we pick our P-value from a larger set of P-values, or change our alpha based on the given data, we can invalidate our tests in a spectacular fashion. But the above points are just part of Neyman–Pearson hypothesis-testing theory and entail no mistake.
The difficulty of creating clear and accurate verbal descriptions of frequentist logic is yet another argument (among others2) for teaching the comparatively straightforward logic of Bayesian statistics, even if one argues that the latter should be calibrated via frequency calculations.1,,11,12
Multiple-comparisons problems are real—and common
In the late 20th century, a sharp division emerged between epidemiologists who condemned all multiple-comparison procedures and statisticians who simply carried on with conventional methods such as Bonferroni adjustments. Both sides made legitimate criticisms of the other's stance, but offered unsatisfactory solutions in return. These issues were brought to the fore by an extended debate in the 1990s.13–21
Suppose one were focused on estimating a single effect, as in a systematic review of β-carotene intake and lung cancer. Some epidemiologists argued that, in principle, it would be nonsensical to worry about what else was examined in the studies, and thus denied the need for multiple-comparison procedures.13,,16 After all, the argument went, the observed association could not depend on whether one looked at other associations.
In reality, careful study of other associations in the data could lead to alterations in confounding control, and in this way alter the estimated β-carotene effect. Furthermore, if one saw the relation of smoking to lung cancer was weak in one of the data sets, it might suggest serious error in the study and reduce the credibility of the estimated β-carotene effect from that study. Continued reasoning along these lines could lead one to employ methods for simultaneous estimation of effects such as shrinkage or Bayes procedures, even if one were ultimately concerned with just one effect and considered the rest only as potential confounders.22,,23
More telling is that the argument against considering whether multiple comparisons were done assumes that what is reported in the literature is an unbiased sample of all that was examined in all the studies. This assumption ignores the selection biases generated by typical uses of conventional statistics.15–17,,22,24–26 The classic such problem is publication bias. A study team with many possible comparisons might present details only for ‘interesting’ findings. In particular, it appears that study teams more often publish results on associations if they are ‘statistically significant’ (P < 0.05)25,,26 unless there are countervailing forces affecting publication decisions.27 If the P-value is a factor in this selection process, the result is a bias away from the null among the results available for inclusion in reviews, whether one considers results across studies (as in meta-analysis) or results within a study (as in genome scans).26 This bias is of special concern in studies that collect data on many variables, whether lifestyle, occupational or genetic.
Publication bias can thus be seen as a variant of the same selection problem that arises in multiple comparisons. Furthermore, even if one focuses on a single association across studies, the publication-bias problem would be aggravated by conventional multiple-comparison adjustments within studies if those studies publish only significant associations. Making a Bonferroni or other conventional correction within studies in effect decreases the α-level required for presentation, and thus further reduces (in a biased fashion) the amount and representativeness of the published information available to reviewers. In other words, the conventional cure for multiple-comparison problems only makes the underlying problem worse (not unlike more than a few conventional medical cures).
Intuition about such problems may be aided by seeing their relation to regression to the mean. As is well known in screening, if we pick out values from a group based on their extremity (whether ‘extremity’ is based on absolute distance, Z-value or P-value) we will usually find our selections fall back (regress) toward the group mean on the next measurement or study. Further, this regression gets bigger as the initial value gets further from the mean. In typical settings with multiple comparisons, extremely significant associations tend to arise more from extreme errors rather than extreme effects, even if the only source of error is random.14 Regression to the mean increases if one uses a more stringent (extreme) selection cut-off, which is what conventional multiple-comparison adjustment do. Add on study validity problems, which should be of paramount concern in observational settings, and conventional multiple-comparison adjustments look like nothing more than added distortion.16,,20
Defensible methods for association selection
One solution to the reporting problem is to provide detailed information on all associations computable from the data. The general infeasibility of this solution is readily apparent: it would not be unusual for a dietary cohort study to have data on 100 food items, 40 food constituents and 30 disease outcomes, resulting in 120 000 associations if we allow only one version of each association. Occupational and medical database studies can generate even more associations. Even if complete presentation were feasible, it would drastically curtail development of careers dependent on number of publications, (so would not be adopted with enthusiasm), and it would not at all address the need to decide which of these associations is worthy of further study.
Thus, some sort of procedure is needed to pick out associations for further attention. If an association has already been called to attention by previous work, it should easily make the ‘short list’ for reporting, because such previous work needs to be checked and the results of that check need to be reported. But what of the remaining associations, those we consider probably null? As Wakefield emphasizes, the P-value alone (with or without ‘correction’) is deficient for selecting among these probably null candidates: It ignores not only Bayesian parameters (prior probabilities), but ignores frequentist parameters (power and precision) as well. In particular, selection based on conventional multiple testing such as Bonferroni adjustment is analogous to choosing a screening test for disease only to ensure a low frequency of false positives (Type I errors) among the non-diseased (high specificity), without regard to false negatives (Type II errors) or background prevalence (prior probability). It should be no surprise that such a deficient criterion leads to poor selection performance.
Frequentist empirical-Bayes methods and their Bayesian counterparts have been recommended for over three decades as replacements for conventional multiple-testing adjustments and variable selection in epidemiology.14,,22–24,28–33 These methods incorporate all the parameters relevant for selection, and perform demonstrably better than conventional adjustments for the actual purposes of most studies. In particular, conventional methods (like Bonferroni procedures) test a joint hypothesis that all the associations examined are null, under the assumption that false discovery is the worst error one can commit. More often, however, the goal is to identify or rank associations for further pursuit. Some Bayesian and empirical-Bayesian methods rank associations based on a summary of their posterior distribution, such as their posterior means or a posterior probability of exceeding a given size. These approaches seem natural, given that the methods account for anticipated regression to the mean by shrinking conventional estimates toward their prior mean,29–32 although for hypothesis screening it is arguably better to estimate the rankings directly.34
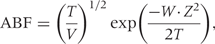
Wakefield studies the performance of this ABF in hypothesis screening (‘fishing expeditions’), especially in relation to ranking by P-values. The method and study seem suitable for the context he describes (ranking a priori exchangeable associations). Nonetheless, I have reservations about the use of Bayes factors (as opposed to rankings based on posterior summaries or direct rank estimation) in the more general setting of only partial exchangeability. At the very least, regressing out sources of non-exchangeability and operating with exchangeable residuals30,,33 would seem essential, but is not addressed by Wakefield's current method (understandably, given his examples do not appear to require such refinements).
P-values = bad Bayes factors
Just after introducing his first ABF (formula 1 above), Wakefield describes a second ABF that provides rankings identical to the P-values from the chosen Wald statistic (the Z-value). It does so by using a different prior for each log odds ratio, assuming the prior variance W proportional to the estimated sampling variance V, i.e. W = K × V for some constant K (which is a data-driven and hence empirical-Bayesian assumption). Wakefield's second approximate Bayes factor suffers from the major problem of P-values that Wakefield's first ABF was developed to address—i.e. its rankings do not depend on power or precision. It also reveals another reason that P-values are a poor choice for ranking: they implicitly correspond to implausible prior distributions.
It is conceivable that our prior variances for log odds ratios would expand as the exposures became more uncommon or the actual log odds ratios moved away from the null. Nonetheless, there is no reason to think that the prior variances would increase at a rate miraculously proportional to the estimated sampling variance. It might be tempting to think so based on confusion of actual measures with their estimates (log odds ratio estimates based on uncommon exposures do tend to have larger sampling variances). But any argument based on the size of the actual effects has to take account of the fact that effect-size rankings depend on the measure of effect. In particular, rankings can be reversed by choosing a difference instead of a ratio.35–37 Complicating matters further, estimated rankings at the far extremes can differ for different effect measures even if we first divide the estimates by their approximate standard errors (Appendix).
Conclusion
The emergence of genetic epidemiology, with its staggering number of associations to explore, has brought multiple-inference concepts into the mainstream of epidemiology and biostatistics.1,,12 It is thus time to recognize of the extent of multiple-comparison problems in everyday epidemiology and deploy modern methods toward their resolution.
Acknowledgements
The author is grateful to Charles Poole, Katherine Hoggatt and Jon Wakefield for helpful comments.
Conflict of interest: None declared.
References
Appendix
Different effect measures can provide different rankings of effect size.35–37 Dividing the estimated measures by their standard errors does not always remove this phenomenon and can even cause it. To see the latter possibility, compare the simple numerical examples in Table A1. Both the risk difference and the risk ratio increase going from panel 1 to panel 2, as does the Z-score (null Wald statistic) from the risk difference. But the Z-score from the log risk ratio declines. The same possibility exists for rare diseases and when using rate ratios or odds ratios in place of risk ratios. These examples reflect the fact that different asymptotic statistics are not equivalent except in neighbourhoods of the test hypothesis.38,,39 Other examples can be constructed in which the estimates agree with their own Z-score rankings, but the rankings differ for the different measures and their Z-scores.
Hypothetical cohort example in which changing to the Z-score (estimate over standard error) reverses the ranking when using the risk ratio but not when using the risk difference, when standard errors are computed using conventional large-sample formulas40, p. 249
| Exposed | Unexposed | Exposed | Unexposed | ||
|---|---|---|---|---|---|
| Cases | 10 | 4 | Cases | 10 | 2 |
| Total | 20 | 80 | Total | 20 | 80 |
| Exposed | Unexposed | Exposed | Unexposed | ||
|---|---|---|---|---|---|
| Cases | 10 | 4 | Cases | 10 | 2 |
| Total | 20 | 80 | Total | 20 | 80 |
RD, Za 0.450, 3.93 0.475, 4.20
RR, Zb 10, 4.12 20, 4.00
aRD = Risk difference estimate, Z = RD divided by standard error.
bRR = Risk ratio estimate, Z = ln(RR) divided by standard error.
Hypothetical cohort example in which changing to the Z-score (estimate over standard error) reverses the ranking when using the risk ratio but not when using the risk difference, when standard errors are computed using conventional large-sample formulas40, p. 249
| Exposed | Unexposed | Exposed | Unexposed | ||
|---|---|---|---|---|---|
| Cases | 10 | 4 | Cases | 10 | 2 |
| Total | 20 | 80 | Total | 20 | 80 |
| Exposed | Unexposed | Exposed | Unexposed | ||
|---|---|---|---|---|---|
| Cases | 10 | 4 | Cases | 10 | 2 |
| Total | 20 | 80 | Total | 20 | 80 |
RD, Za 0.450, 3.93 0.475, 4.20
RR, Zb 10, 4.12 20, 4.00
aRD = Risk difference estimate, Z = RD divided by standard error.
bRR = Risk ratio estimate, Z = ln(RR) divided by standard error.

