Article Text

Statistics from Altmetric.com
Until recently, atherosclerosis was thought of as a degenerative, slowly progressive disease, predominantly affecting the elderly, and causing symptoms through its mechanical effects on blood flow, particularly in the small calibre arteries supplying the myocardium and brain. Thus the approach to treatment has traditionally been surgical and focused on the largest and most visible or symptomatic lesions, coupled with a somewhat nihilistic belief that there was little likelihood of medical management affecting such a longstanding “end stage” process. However, recent research into the cellular and molecular events underlying the development and progression of atherosclerosis, prompted by careful descriptive studies of the underlying pathology, has shown that atherosclerosis is a dynamic, inflammatory process that is eminently modifiable. Support for this view comes from clinical trials of lipid lowering agents, particularly the “statins”, which have shown only minor effects on the size of existing lesions, but major reductions in clinical events caused by plaque rupture, implying a beneficial stabilising effect on plaque composition. This calls for a change from a quantitative (how many and how tight are the stenoses?) to a more qualitative (how active are the plaques we cannot see?) approach to atherosclerosis. Also, a better understanding of the molecular and cellular basis of atherosclerosis will inevitably lead to the design of better diagnostic and therapeutic approaches. The purpose of this review is to summarise current understanding of the pathogenesis and progression of atherosclerosis with particular reference to potential new diagnostic or therapeutic approaches.
The atherosclerotic plaque
Atherosclerosis begins as a subendothelial accumulation of lipid laden, monocyte derived foam cells and associated T cells which form a non-stenotic fatty streak. With progression, the lesions take the form of an acellular core of cholesterol esters bounded by an endothelialised fibrous cap containing vascular smooth muscle cells (VSMC) and inflammatory cells, predominantly macrophages with some T cells and mast cells, which tend to accumulate at the shoulder regions of the plaque. Also present in advanced lesions are new blood vessels and deposits of calcium hydroxyapatite. Thus atherosclerotic lesions are complex and it is the dynamic interaction between the different components of the plaque that dictates outcome of the disease.
The endothelium
Since the discovery of endothelium dependent relaxation, and its biochemical entity, nitric oxide (NO), it has been recognised that the endothelium plays an important role in vascular biology. Several roles have been ascribed to vascular NO including modulation of tone, inhibition of platelet aggregation, inhibition of VSMC proliferation, and production of potentially destructive free radicals, in particular peroxynitrite. The earliest detectable physiological manifestation of atherosclerosis is reduced production of NO in response to pharmacological or haemodynamic stimuli. This phenomenon is present even in children with hypercholesterolaemia,1 and is consistent with the hypothesis that high circulating concentrations of atherogenic lipoproteins lead to endothelial dysfunction and (by unknown mechanisms) subendothelial lipid accumulation. Importantly, however, it remains unclear whether this manifestation of endothelial dysfunction is a cause or a consequence of lipid accumulation, since fatty streaks are also present from a young age. Interestingly, a number of drugs that are known to influence beneficially the outcome of vascular disease, including statins and angiotensin converting enzyme (ACE) inhibitors, have been shown to improve endothelial function in studies of brachial artery dilatation in response to increased forearm blood flow.
In addition to reduced NO bioavailability, and possibly partly because of it, endothelial cells in atherosclerosis express surface bound molecules (selectins and adhesion molecules) that attract and capture circulating inflammatory cells and facilitate their migration into the subendothelial space. The importance of these molecules in the development of atherosclerosis is demonstrated in mice deficient in molecules such as intercellular adhesion molecule 1 (ICAM 1) and P selectin, which develop smaller lesions with less lipid and fewer inflammatory cells than control mice when fed a high lipid diet. Thus recruitment of inflammatory cells is important for plaque development, but since inflammatory cells do not accumulate in the intima in the absence of lipid, these data suggest that lipid accumulation is the initiating event and that inflammatory cells play a permissive role in lesion progression. The predisposition for atherosclerosis at particular sites in the vascular tree may be influenced by subtle, local effects on endothelial function, particularly shear stress which is known to regulate expression of a number of endothelial cell genes including ICAM 1 and eNOS. It remains to be seen whether new treatments being designed, for example, to enhance deficient NO bioavailability or to reduce adhesion molecule expression will have any impact on atherosclerosis or its consequences in man.
Inflammatory cells
Accumulated subendothelial lipid, particularly if oxidised, exacerbates the local inflammatory reaction and maintains activation of the overlying endothelium. This results in continued expression of selectins and adhesion molecules and also expression of chemokines, in particular monocyte chemoattractant proteins-1 (MCP-1). Chemokines are proinflammatory cytokines that function in leucocyte chemoattraction and activation. Atheroma prone mice lacking MCP-1 develop smaller atherosclerotic lesions than mice expressing MCP-1, providing further evidence that inflammatory cells play an important part in lesion development. Once captured, the inflammatory cells migrate into the subendothelial space where, under the influence of local chemokines, they become activated. The monocytes mature into macrophages and express the necessary scavenger receptor to ingest modified lipids and become macrophage foam cells. The predominant role of the macrophage in atherosclerosis is to ingest and dispose of atherogenic lipids. However, activated macrophages and T cells also express a variety of proinflammatory cytokines and growth factors that contribute to the evolution of the plaque. The progression of an atherosclerotic plaque is best understood in terms of the dynamic interaction between a subendothelial inflammatory stimulus and the local reactive “wound healing” response of surrounding VSMCs.
Vascular smooth muscle cells
Medial VSMCs contain large amounts of contractile proteins since their predominant role is to maintain vascular tone. This “contractile” VSMC phenotype is maintained particularly by the influence of extracellular proteins in the media on VSMC surface integrins. However, if medial VSMCs are taken out of this environment, for example into cell culture, they undergo a change in phenotype characterised by a reduction in content of contractile proteins and an increase in synthetic organelles. VSMCs undergo a similar change when they move from the media to the intima in vivo. This change in phenotype from “contractile” to “synthetic” was once thought of as being a key initiating event in the pathogenesis of an atherosclerotic lesion.2 However, recent studies have shown a remarkable similarity in gene expression between intimal VSMCs in atherosclerosis and VSMCs in the early developing blood vessel,3 consistent with the view that intimal VSMCs are more likely to be performing a reparative than a permissive role in atherosclerosis. In adopting a “repair” phenotype VSMCs express the proteinases that are required to break down the surrounding basement membrane to facilitate their migration to the site of injury; they produce growth factors that facilitate their proliferation at the site of injury, and they produce the necessary matrix proteins, in particular collagens and elastin, to repair the vessel. Indeed, expression of this repertoire of genes is essential for the formation of a fibrous cap over the lipid core of an atherosclerotic plaque. Since the fibrous cap separates the highly thrombogenic lipid rich core from circulating platelets and proteins of the coagulation cascade and confers structural stability to an atherosclerotic lesion, and since the VSMC is the only cell capable of synthesising the cap, it follows that VSMCs play a pivotal role in maintaining plaque stability and protecting against plaque rupture and consequent thrombosis.4
Cellular interactions and plaque stability
Atherosclerotic lesions are ubiquitous in most adults in the developed world, but they are largely asymptomatic. There are two mechanisms by which atherosclerosis leads to symptoms. If the lesion becomes sufficiently large to restrict blood flow, such that nutrient supply cannot meet demand, then tissue ischaemia will occur, as in chronic stable angina pectoris. However, plaque growth does not always lead to lumen stenosis, since atherosclerotic arteries can remodel to accommodate the expanding atherosclerotic lesion while still maintaining a normal or near normal lumen diameter.5 Thus large atherosclerotic lesions may be, and often are, clinically silent.
If the lesion ruptures or erodes platelets rapidly accumulate and intravascular thrombosis can occur, leading to the acute coronary syndromes of unstable angina and myocardial infarction. Plaques with a large lipid pool and a thin fibrous cap are much more prone to rupture than those with a thick cap, partly because a thick fibrous cap is more able to resist local mechanical stresses. However, the most important determinant of plaque stability is the composition of the fibrous cap, in that a preponderance of inflammatory cells and a relative paucity of VSMCs leads to plaque rupture.6
There are several mechanisms by which inflammatory cells can weaken the fibrous cap. Firstly, they affect matrix protein turnover by producing specific metalloproteinases that degrade matrix proteins in the cap4 and proinflammatory cytokines, in particular interferon γ (INF γ), that inhibit VSMC proliferation and collagen synthesis. Secondly, they secrete inflammatory cytokines, in particular interleukin 1β, tumour necrosis factor α (TNF α) and INF γ, that are synergistically cytotoxic for VSMCs. Thirdly, it has recently been shown that activated macrophages can induce VSMC apoptosis (programmed cell death) by direct cell-cell contact. All this is further compounded by the phenotype of the VSMCs within the fibrous cap of a mature plaque which have a reduced ability to proliferate and an enhanced susceptibility to apoptosis.7 ,8 Thus inflammatory cells can destroy the fabric of the fibrous cap and resident VSMCs are poorly equipped to compensate. Importantly, these features can be and often are present in small, haemodynamically insignificant atherosclerotic plaques that are clinically silent and angiographically invisible. Thus plaque composition is far more important than plaque size in determining outcome.
Consequences of plaque rupture
Rupture or erosion of the fibrous cap exposes the highly thrombogenic collagenous matrix and lipid core to the circulation and leads inevitably to platelet accumulation and activation. This, in turn, leads to fibrin deposition, thrombus formation and, at its most extreme, vessel occlusion. However, vessel occlusion is not inevitable, and it is now clear that episodes of silent, subclinical plaque rupture occur frequently in patients with atherosclerosis. In one study up to 70% of plaques causing high grade stenosis had evidence of previous plaque rupture and repair in the absence of vessel occlusion or a corresponding clinical event.9 These episodes of non-occlusive plaque rupture induce recruitment of new VSMCs under the influence of mitogens, in particular platelet derived growth factor and thrombin10 in the thrombus. Thrombus also contains large quantities of transforming growth factor β which is a potent stimulator of VSMC matrix synthesis. These factors therefore drive formation of a new fibrous cap over the thrombus, thereby increasing the size of the lesion (fig 1). Thus the size of atherosclerotic lesions increases as a consequence of repeated episodes of rupture and repair. The chief implication of this is that pharmacological inhibition of silent plaque rupture would be expected to reduce progression of atherosclerotic lesions. It also serves to explain and emphasise the value of antiplatelet drugs in atherosclerosis.
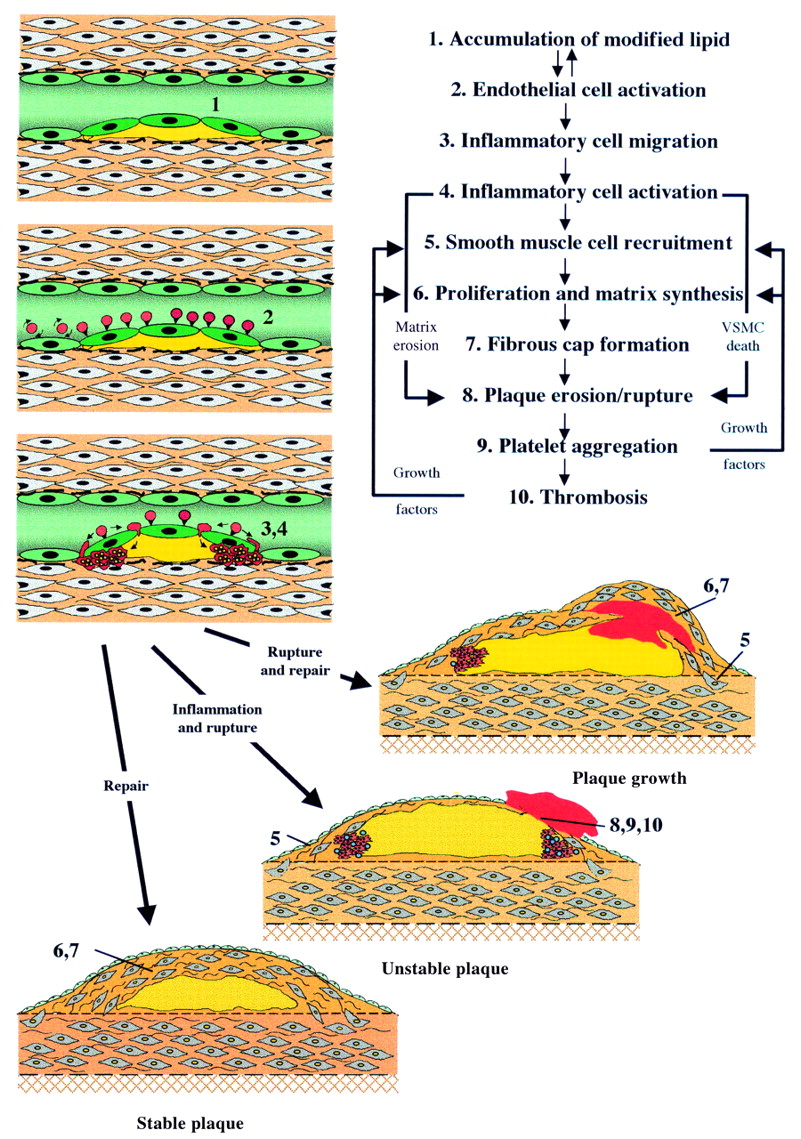
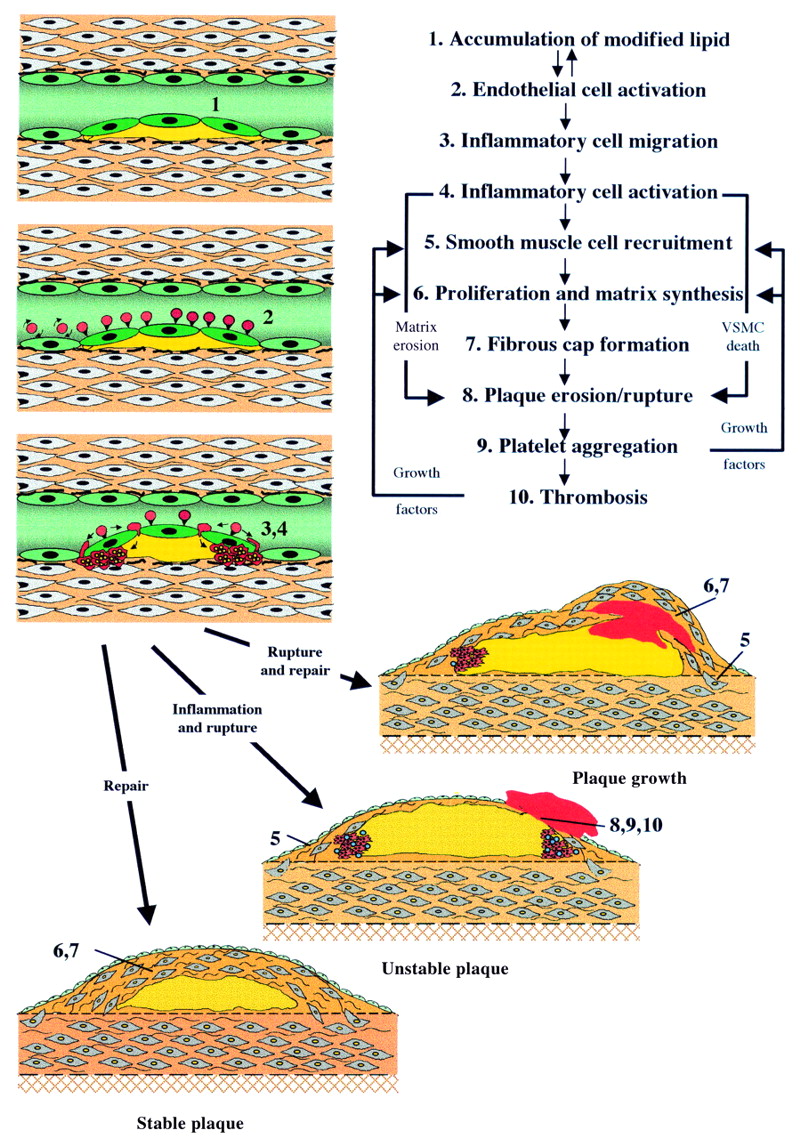{kind=link}
Cellular interactions in the development and progression of atherosclerosis. VSMC, vascular smooth muscle cells.
Diagnostic implications of plaque biology
Imaging
The mainstay of clinical diagnosis of coronary disease is the coronary angiogram. Angiography can only detect lesions that impinge significantly on the lumen, however, and provide little or no information on the composition of a stenotic lesion. Since it is composition rather than size that determines the likelihood of plaque rupture, it follows that angiography is likely to be a poor predictor of clinical events. Thus, Falk and colleagues11 showed that most of the lesions that cause myocardial infarction produce less than a 50% stenosis. This important observation explains why myocardial infarction occurs so commonly in patients who have experienced no previous symptoms and emphasises why better diagnostic tools are required. Thus, despite its continuing pivotal role in evaluation and management of symptomatic coronary disease, angiography has little to offer in risk prediction or therapeutic monitoring in the asymptomatic population. The same is true for intravascular ultrasound, even though it provides much more information than angiography on the extent and composition of targeted plaques. Ideally, what is required is an imaging technique that can be used repeatedly to identify and monitor asymptomatic atherosclerosis.
The extent of coronary calcification, as quantified by electron beam computed tomography, has been shown to predict clinical events, and an absence of calcification can be taken to indicate an absence of significant coronary disease. The precise relation between calcification and plaque progression remains to be determined, however, and monitoring calcification will only be of value if it proves to be reversible as indicated by recent studies.12
The most promising emerging imaging techniques are magnetic resonance imaging (MRI) and radionucleide based techniques, in particular positron emission tomography (PET). MRI can differentiate some plaque components in animal models and human vessels.13 However, image resolution and movement artefact remain substantial obstacles to the use of MRI to monitor coronary disease and while MRI may provide fine anatomical detail, it is unlikely to provide information on inflammatory activity within plaques. In contrast, PET provides little anatomical information but offers the potential to measure and monitor plaque inflammatory cell content and activity. Early animal studies using [18F]FDG-PET to measure plaque metabolic activity have been encouraging, but as with MRI, resolution is a major obstacle to imaging coronary atheroma in man. Despite these obstacles, it seems inevitable that non-angiographic imaging of atherosclerosis will be developed in the foreseeable future.
Risk factor evaluation
The realisation that atherosclerosis is essentially an inflammatory process has prompted the search for measurable biochemical markers of plaque inflammation, some of which are non-specific, such as serum amyloid A (SAA), C reactive protein (CRP) and TNF α, while others, such as ICAM 1 and VCAM 1, are thought to be more specific for vascular inflammation. Most prominent of these is the use of highly sensitive assays to measure low concentrations of CRP—that is, below the limit of detection of assays used in routine clinical practice—which have shown a strong correlation between CRP and risk of myocardial infarction and stroke.14 Furthermore, those patients with highest CRP concentrations (albeit within the conventional normal range) derived most benefit from prophylactic aspirin treatment. Also, benefit derived from pravastatin treatment in the CARE study was associated with a reduction in CRP and SAA concentrations. It is likely that in the near future biochemical measures of inflammation, in combination with measurement of conventional risk factors, will be used to guide selection of patients at highest risk for primary prevention treatment and to monitor their progress. Also, it is likely that a better understanding of the molecular interactions that dictate plaque instability will lead to the discovery of more specific circulating markers of disease activity.
Therapeutic implications of plaque biology
Angiographic studies have shown that effective lipid lowering with statins reduces the incidence of new lesion formation and produces a significant, but haemodynamically unimportant (0.04–0.07 mm), improvement in established stenoses.15 Importantly, however, they also reduce the rate of progression of pre-existing lesions and the number of new vessel occlusions. Both of these beneficial effects are likely to be due to prevention of plaque rupture since, as discussed above, lesions grow by repeated episodes of subclinical rupture and repair, and silent occlusion arises when plaque rupture and thrombosis occur in the context of a well collateralised myocardial circulation such that no significant ischaemia results from the occlusion. Despite the modest effects of statins on size of pre-existing lesions, several outcome studies, both in primary and secondary prevention settings, have shown a substantial (30–40%) reduction in cardiovascular events in the statin treated groups.16-18 Taking the angiographic and outcome data together, these results argue strongly that statins stabilise plaques.
The balance of atherosclerosis
Atherosclerosis is a dynamic balance between the destructive influence of inflammatory cells and the reactive, stabilising effects of VSMCs. The balance is biased in favour of plaque rupture by factors such as high low density lipoprotein (LDL) cholesterol, lipid peroxidation and, probably, genetic variability in the inflammatory molecules involved. For example, there is a correlation between plaque progression and a polymorphism in the stromelysin-1 gene promoter. Also, it is entirely plausible that infective agents, in particularChlamydia pneumoniae, which can be found in plaque macrophages, may exacerbate the inflammatory process and tip the balance in favour of plaque rupture; this hypothesis is currently being tested in clinical trials.
In contrast, the balance will be biased towards repair and stability by a reduction in plaque inflammation. Lipid lowering, by whatever means, is associated with a reduction in clinical events and animal studies have shown dramatic reductions in plaque inflammatory cell content during statin treatment, even in the absence of lipid lowering.19 These observations, coupled with a greater benefit from pravastatin treatment than was predicted by the achieved reduction in LDL cholesterol in the West of Scotland coronary prevention study,16 have led to the suggestion that some statins may exert effects on plaque stability which are additional to and independent of lipid lowering. Laboratory studies have shown that statins can exert direct effects on endothelial cell function, inflammatory cell activity, VSMC proliferation, platelet aggregation, and thrombus formation.20 ,21 Since the effects of different stains are not equivalent, it has been suggested that some may afford more or less protection than others for an equivalent lipid lowering effect. Such observations argue strongly for robust outcome studies to prove overall efficacy and safety.
Understanding of the non-lipid associated events in atherogenesis raises the prospect of developing drugs targeted at specific events in its pathogenesis which might act synergistically with lipid lowering drugs to enhance plaque stability. Possible targets include endothelial NO and adhesion molecule production, the matrix metalloproteinases, inflammatory cytokines and their receptors, and angiogenesis, since mice lacking specific angiogenic factors develop smaller atherosclerotic lesions than controls. If oxidised lipids are the major stimulus for atherogenesis, then antioxidants, such as vitamin E, might be expected to reduce the inflammatory drive in atherosclerosis and thereby promote plaque stability. However, despite early promise, recent large scale studies have cast doubt on the integrity of this hypothesis.
Atherosclerosis is not thought to represent an inflammatory reaction to the subendothelial accumulation of modified lipid
Atherosclerosis is invariably associated with abnormal endothelial cell function. Vascular smooth muscle cells are the only cells capable of protecting against plaque rupture and its consequences
The outcome of atherosclerosis is determined much more by plaque composition than plaque size
Atherosclerotic lesions frequently enlarge as a consequence of repeated subclinical episodes of rupture and repair. The only logical conclusion to be drawn from the angiographic and outcome studies of statin treatment is that statin treatment stabilises atherosclerotic lesions
Atherosclerosis is a dynamic process capable of being modified
In addition to reducing inflammation, stimulation of the VSMC repair process should result in increased stability. For example, balloon angioplasty stimulates a vigorous VSMC repair response. This may contribute to restenosis and the re-emergence of angina, but the resulting lesion is always fibrotic and stable and rarely if ever precipitates an acute coronary event, even when the original target lesion was unstable. It is feasible therefore that better understanding of the molecular regulators of VSMC behaviour may lead to drug treatments aimed at enhancing fibrous cap formation. Modulators of TGF β activity may have a particularly important role to play in this context.
Summary
Atherosclerosis is a dynamic balance between lipid driven inflammatory cells and their cytokines within the substance of the plaque, and the natural stabilising properties of the surrounding VSMCs. That plaque composition is much more important than size is illustrated by clinical and laboratory studies with statins which have little effect on plaque size yet have a major influence on plaque composition and clinical outcome. In future, cardiologists will need to re-focus their attention away from angiographic appearances in symptomatic patients towards potential measures of inflammatory atherosclerotic activity in asymptomatic patients with subclinical disease. Our evolving knowledge of the cellular and molecular interactions that lead to plaque development and progression is likely to lead to novel imaging strategies and novel biochemical measures of disease progression, and potentially also to the development of drugs aimed specifically at stabilising atherosclerotic lesions. That this is achievable has already been demonstrated.
Acknowledgments
PLW is the British Heart Foundation Professor of Cardiovascular Medicine.
References
Supplementary materials
- Additional references for: Atherogenesis: current understanding of the causes of atheroma. Weissberg PL.Heart 2000;83:247�52.
Nakashima Y, Raines E, Plump A, et al. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler Thromb Vasc Biol 1998;18:842�51.
Gosling J, Slaymaker S, Gu L, et al. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest 1999;103:773�8.
Campbell GR, Campbell JH. Smooth muscle phenotypic changes in arterial wall homeostasis: implications for the pathogenesis of atherosclerosis. Exp Mol Pathol 1985;42:139�62.
Warner SJ, Friedman GB, Libby P. Immune interferon inhibits proliferation and induces 2'-5'-oligoadenylate synthetase gene expression in human vascular smooth muscle cells. J Clin Invest 1989;83:1174�82.
Amento EP, Ehsani N, Palmer H, et al. Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arterioscler Thromb 1991;11:1223�30.
Geng Y, Wu Q, Muszynski M, et al. Apoptosis of vascular smooth-muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factor-alpha, and interleukin-1-beta. Arterioscler Thromb Vasc Biol 1996;16:19�27.
Boyle J, Bennett M, Proudfoot D, et al. Human monocyte/macrophages induce human vascular smooth muscle cell apoptosis in culture [abstract]. J Pathol 1998;184:A13.
Bennett MR, Littlewood TD, Schwartz SM, et al. Increased sensitivity of human vascular smooth muscle cells from atherosclerotic plaque to p53-mediated apoptosis. Circ Res 1997;81:591�9.
Grainger DJ, Wakefield L, Bethell HW, et al. Release and activation of platelet latent TGF-beta in blood clots during dissolution with plasmin. Nat Med 1995;1:932�7.
Budoff MJ, Shavelle DM, Lamont DH, et al. Usefulness of electron beam computed tomography scanning for distinguishing ischemic from nonischemic cardiomyopathy. J Am Coll Cardiol 1998;32:1173�8.
Vallabhajosula S. Radioisotopic imaging of atheroma. In: Fuster V, ed. The vulnerable atherosclerotic plaque: understanding, identification, and modification. New York: Futura Publishing; 1999:213�29.
Ridker PM, Rifai N, Pfeffer MA, et al. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events (CARE) investigators. Circulation 1998;98:839�44.
Pitt B, Mancini GB, Ellis SG, et al. Pravastatin limitation of atherosclerosis in the coronary arteries (PLAC I): reduction in atherosclerosis progression and clinical events. PLAC I investigation. J Am Coll Cardiol 1995;26:1133�9.
Jukema JW, Bruschke AV, van Boven AJ, et al. Effects of lipid lowering by pravastatin on progression and regression of coronary artery disease in symptomatic men with normal to moderately elevated serum cholesterol levels. The regression growth evaluation statin study (REGRESS). Circulation 1995;91:2528�40.
Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events trial investigators [see comments]. N Engl J Med 1996;335:1001�9.
Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas coronary atherosclerosis prevention study [see comments]. JAMA 1998;279:1615�22.
Ye S, Eriksson P, Hamsten A, et al. Progression of coronary atherosclerosis is associated with a common genetic variant of the human stromelysin-1 promoter which results in reduced gene expression. J Biol Chem 1996;271:13055�60.
Kol A, Sukhova GK, Lichtman AH, et al. Chlamydial heat shock protein 60 localizes in human atheroma and regulates macrophage tumor necrosis factor-alpha and matrix metalloproteinase expression. Circulation 1998;98:300�7.
Shiomi M, Ito T, Tsukada T, et al. Reduction of serum-cholesterol levels alters lesional composition of atherosclerotic plaques � effect of pravastatin sodium on atherosclerosis in mature WHHL rabbits. Arterioscler Thromb Vasc Biol 1995;15:1938�44.
Katznelson S, Wang XM, Chia D, et al. The inhibitory effects of pravastatin on natural killer cell activity in vivo and on cytotoxic T lymphocyte activity in vitro. J Heart Lung Transplant 1998;17:335�40.
Negre-Aminoux P, van Vliet A, van Erck M, et al. Inhibition of proliferation of human smooth muscle cells by various HMG-CoA reductase inhibitors: comparison with other human cell types. Biochem Biophys Acta 1997;1345:259�68.
Lacoste L, Lam JY, Hung J, et al. Hyperlipidemia and coronary disease. Correction of the increased thrombogenic potential with cholesterol reduction [see comments]. Circulation 1995;92:3172�7.
Stephens NG, Parsons A, Schofield PM, et al. Randomised controlled trial of vitamin E in patients with coronary disease: Cambridge heart antioxidant study (CHAOS) [see comments]. Lancet 1996;347:781�6.
GISSI Investigators. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-prevenzione trial. Gruppo Italiano per lo studio della sopravvivenza nell'Infarto miocardico [see comments]. Lancet 1999;354:447�55.