Article Text

Abstract
Background and Aims: Epidemiology studies have shown that cardiovascular (CV) disease is primarily responsible for the mortality associated with increased pulmonary environmental particle (PM10) exposure. The mechanisms involved in PM10 mediated CV effects are unknown although changes in plasma viscosity and in the homoeostasis of blood coagulation have been implicated. It was hypothesised that PM10 exposure would result in an inflammatory response and enhance the activation of the extrinsic coagulation mechanisms in pulmonary and vascular cells in culture.
Methods: Primary human monocyte derived macrophages and human umbilical cord vein endothelial, human alveolar type II epithelial (A549), and human bronchial epithelial (16HBE) cells were tested for their inflammatory and procoagulant response to PM10 exposure. IL-8, tissue factor (TF), and tissue plasminogen activator (tPA) gene expression and protein release, and coagulation enhancing ability of culture media were determined 6 and 24 hours following exposure.
Results: The culture media from macrophages and 16HBE bronchial epithelial cells, but not A549 cells, exposed to PM10 had an enhanced ability to cause clotting. Furthermore, H2O2 also increased the clotting activity. Apoptosis was significantly increased in macrophages exposed to PM10 and LPS as shown by annexin V binding. TF gene expression was enhanced in macrophages exposed to PM10, and HUVEC tissue factor and tPA gene and protein expression were inhibited.
Conclusions: These data indicate that PM10 has the ability to alter macrophage, epithelial, and endothelial cell function to favour blood coagulation via activation of the extrinsic pathway and inhibition of fibrinolysis pathways.
- ARDS, adult respiratory distress syndrome
- COPD, chronic obstructive pulmonary disease
- CV, cardiovascular
- HBE, human bronchial epithelial
- HUVEC, human umbilical vein endothelial cell
- IL, interleukin
- LPS, lipopolysaccharide
- PAI, plasminogen activator inhibitor
- PM, particulate matter
- TF, tissue factor
- TNF, tumour necrosis factor
- tPA, tissue plasminogen activator
- PM10
- cardiovascular
- thrombosis
- inflammation
Statistics from Altmetric.com
- ARDS, adult respiratory distress syndrome
- COPD, chronic obstructive pulmonary disease
- CV, cardiovascular
- HBE, human bronchial epithelial
- HUVEC, human umbilical vein endothelial cell
- IL, interleukin
- LPS, lipopolysaccharide
- PAI, plasminogen activator inhibitor
- PM, particulate matter
- TF, tissue factor
- TNF, tumour necrosis factor
- tPA, tissue plasminogen activator
There is a strong association between increases in environmental particles (PM2.5 and PM10) and adverse effects on pulmonary and cardiovascular health. Pulmonary effects include increased frequency of exacerbations of asthma and chronic obstructive pulmonary disease (COPD), and increased mortality.1 Lung cancer deaths are also increased in areas of high particulate pollution.2 At concentrations commonly attained in US and European urban areas,1,3,4 increased levels of particles are also associated with an increased prevalence in cardiovascular exacerbations.2,4,5 This cardiovascular impact has been suggested to be at least as great as that on respiratory disease despite the latter having a greater relative risk. In one study the respiratory and cardiovascular mortality associated with particulate air pollution increased by 0.54% and 0.69% respectively with a rise of 10 μg/m3 in PM10.6 Pope and colleagues3 found that a 10 μg/m3 increase in PM10 concentration was associated with a 1% increase in daily mortality, a 3.4% increase in respiratory related mortality, and a 1.8% increase in cardiovascular related mortality. A comparable increase in PM2.5 was associated with a rise in total and respiratory/cardiovascular mortality of 4% and 6% respectively.2
The mechanism of the adverse cardiovascular effects caused by inhalation of particulate air pollution remains unclear. We have hypothesised that ultrafine particle mediated lung inflammation could lead to a systemic increase in blood coagulability.7 This has been supported by studies during a pollution episode.8,9 Proinflammatory properties of PM10 have been identified in vitro10,11 and in vivo in healthy volunteers.12 There is evidence that C reactive protein is increased in response to particulate air pollution,9 and there are clear links between inflammation and atherothrombosis. It has been suggested that inhaled particles may induce a prothrombotic state, through increased plasma viscosity,8 and increased plasma fibrinogen and factor VII concentrations.9,12,13 Furthermore, experimental studies have supported an effect of PM10 on the development and progression of atherosclerotic plaque formation and rupture, a process which is central to the pathogenesis of acute arterial thrombotic events.14
Identifying pathogenetic mechanisms in models employing healthy animals or in experiments with normal human subjects has been difficult. Stimulation of bone marrow myelopoiesis by particle inhalation has been reported in animal studies,15,16 confirming a remote systemic effect.13 However, in experimental animals blood hypercoagulability has not been noted.
One plausible mechanism to explain particle mediated hypercoagulability is the induction of tissue factor (TF). TF is a cell surface glycoprotein and is a receptor and cofactor for activated factor VII (FVIIa). The formation of a TF:FVIIa complex initiates the extrinsic blood coagulation pathway. This is regarded now as the key route to thrombin generation in vivo and is activated in tissue damage and inflammation. TF expression can be induced in a number of cell types in response to injury or inflammatory mediators,17 including vascular endothelial18 and epithelial cells19 and macrophages.20 TF:FVIIa expression on the cell surface is regulated by TF gene expression and the availability of anionic phospholipids21 such as those present on the surface of apoptotic cells.22 Anionic phospholipids themselves promote coagulation by acting as a template for the assembly of the tenase and prothrombinase complexes, which facilitates the generation of thrombin.23
Inhibition of the fibrinolytic pathway results in a prothrombotic state, also. Efficient thrombus lysis requires the activation of plasminogen to plasmin. This is principally through the action of tissue plasminogen activator (tPA). The activity of tPA is regulated by plasminogen activator inhibitor 1 (PAI-1). Alterations in the balance between tPA and PAI-1 have been associated with inflammatory states such as pneumonia,24 sepsis,25 and adult respiratory distress syndrome (ARDS).26
Vascular endothelial cells have an enhanced procoagulant activity and impaired fibrinolytic ability in lung conditions such as sepsis, acute lung injury,27 and the post-operative state.28 Therefore we hypothesise that generation of procoagulant activity and/or inhibition of fibrinolysis in the lung and in the pulmonary microvasculature may play a role in the cardiovascular effects of PM10.
In order to explore the possibility that PM10 can induce prothrombotic changes in human macrophages, endothelial cell and pulmonary epithelial cell prothrombotic and fibrinolytic responses to PM10 exposure were investigated.
MATERIALS AND METHODS
Reagents
All chemicals used in this study were obtained from Sigma Chemical Co., Poole, UK. Cell culture media and reagents were obtained from Gibco-BRL, Paisley, UK. Human recombinant TNFα (R&D Systems, Abingdon, UK) was stored at −80°C at a concentration of 10 μg/ml in sterile distilled water and was diluted in culture media to 10 ng/ml for use. Hydrogen peroxide (H2O2) was prepared in a stock solution of 2 mM in phosphate buffered saline (PBS) and experiments were carried out at a final concentration of 200 μM. Lipopolysaccharide (LPS) was prepared at 1 mg/ml in dH2O, stored at –80°C, and used in cell exposures at a final concentration of either 1 or 10 μg/ml. Staurosporine was stored at –20°C at 10 μg/ml and used at a final concentration of 10 ng/ml.
Cell culture
Human lung type II alveolar-like epithelial cells (A549) and human bronchial epithelial cells (16HBE) were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) supplemented with 10% heat inactivated fetal calf serum (FCS), L-glutamine (2 mM), and penicillin/streptomycin solution in 5% CO2 at 37°C as previously described.10
Human peripheral blood monocyte derived macrophages were isolated as previously described.11 Briefly, 40 ml whole blood was collected from healthy volunteers into 50 ml Falcon tubes containing sodium citrate anticoagulant and sedimented with 6% dextran 500. The leucocyte rich layer was carefully removed and the mononuclear fraction isolated using an isotonic discontinuous PBS/Percoll gradient (Amersham Pharmacia Biotech, Bucks, UK) by centrifugation. The monocytes were collected from the Percoll gradient, washed in PBS, and were seeded into 24 well plates at a density of 4 million cells per ml in RPMI medium containing L-glutamine (2 mM) and penicillin/streptomycin solution without serum, and incubated at 37°C in 5% CO2. After one hour, the medium was removed along with non-adherent cells, the monocytes washed, and the medium replaced with RPMI medium containing 10% autologous serum obtained from the donor during the cell isolation process. The monocytes were matured for five days into macrophages with media changes every two days.
Human umbilical vein endothelial cells (HUVECs) were isolated as previously described.29 Briefly, fresh human umbilical veins were washed with PBS (without Ca2+ and Mg2+), filled with pre-warmed 0.05% collagenase solution, and incubated at 37°C for 6 minutes. Thereafter the veins were perfused with PBS to harvest cells. Cells were collected from the perfusate by centrifugation at 1000 rpm for 5 minutes; isolated, pelleted cells were resuspended and cultured in EGM endothelial cell medium (Clonetics, Wokingham, UK) containing all enclosed supplements (including 2% v/v fetal bovine serum but without gentamicin), and grown in 175 cm3 culture flasks.
Cell exposures
Cells were plated at a density of 0.15×106 cells/well in 24 well plates and grown overnight until 80% confluent. The medium was removed from the cultures and was replaced with serum free medium for a further 24 hours incubation. Test compounds were added for specified times (see results) in serum free medium.
Particles
Carbon black, which was used in the determination of PM10 concentration, was obtained from Degussa Ltd, Frankfurt, Germany.
Particulate matter, of which 50% was <10 μm diameter (PM10) was obtained, quantified, and used as previously described.10 Briefly, PM10 particles were collected on filters in the tapered element oscillating micro-balance of the Marylebone and Bloomsbury, London, monitoring sites of the United Kingdom enhanced urban network. To deliver PM10 into solution, filters were cut in half and each was sonicated into 1 ml of PBS for 1 minute, vortex mixed, and the half-filter removed. Particle concentration was determined by spectrophotometric comparison with a standard curve of turbidity at 340 nm. As well as estimating the dose of PM10, the use of carbon black in the standard curve allowed the dose to be standardised relative to a toxicologically important component of PM10. All PM10 cell exposures were carried out at a concentration of 50 μg/ml PM10 in culture medium unless indicated otherwise in results.
Effects of cell culture supernatants on the recalcification time of citrated human plasma
A 100 μl aliquot of culture supernatant was added to the same volume of normal human plasma pool and mixed at 37°C. A 100 μl aliquot of 25 mM calcium chloride was added, and the time measured for a visible clot to form while manually tipping the tube periodically from the waterbath was noted. Manchester Reagent rabbit brain thromboplastin was used as a positive control and phosphate buffered saline as a negative control. Control experiments were also performed which excluded any direct effect of particles on clotting times.
Reverse transcriptase polymerase chain reaction (RT-PCR)
After exposure to test materials, RNA was isolated from PBS washed cells using the TRIzol reagent (Gibco-BRL, Paisley, Scotland), according to the manufacturer’s instructions, and dissolved in 50 μl diethylpyrocarbonate (DEPC) treated water. Moloney murine leukemia virus reverse transcriptase (M-MLV RT) (Promega, Southampton, UK) was used to transcribe cDNA from 2 μg mRNA according to the manufacturer’s instructions. The genes investigated were the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH), interleukin-8 (IL-8), tissue factor, and tPA with the primer oligonucleotides purchased from MWG-Biotech (Milton Keynes, UK). The following primer pairs were used: GAPDH, sense 5′-CCA CCC ATG GCA AAT TCC ATG GCA-3′, and antisense 5′-TCT AGA CGG CAG GTC AGG TCC ACC-3′; IL-8, sense 5′-CGA TGT CAG TGC ATA AAG ACA-3′, and antisense 5′-TGA ATT CTC AGC CCT CTT CAA AAA-3′; tissue factor, sense 5′-CCC TGA ATT TGA CAG TCT CAC C-3′, and antisense 5′-CAC AAT AAA ACT TGC CCA GAA AAA C-3′; and tPA, sense 5′-GTC TGC TGT GCA CCA TCC CCC ATC-3′, and antisense, 5′-TTG TCA TCA ATC TTG AAT CCC ATA-3′. The primers were diluted to 100 pmol/μl with DEPC water. For each PCR reaction 5 μl of reverse transcribed DNA (cDNA) was added directly to a PCR reaction mixture set to a final volume of 50 μl containing 1 × Taq DNA polymerase reaction buffer (Promega, UK), 2.5 mM MgCl2, 0.2 mM dNTP mixture, 1 unit Taq DNA polymerase (Promega, UK), and 1 μM of the appropriate primer pair. The IL-8 PCR conditions were 27 thermal cycles of 94°C for 30 seconds, 60°C for 1 minute, and 68°C for 2 minutes followed by a final extension step of 68°C for 7 minutes, resulting in a product 180 base pairs in size. The conditions for GAPDH were 25 thermal cycles of 94°C for 45 seconds, 60°C for 45 seconds, and 72°C for 1 minute 30 seconds followed by a final extension step of 68°C for 7 minutes, resulting in a product 600 base pairs in size. The conditions for tissue factor were 35 thermal cycles of 94°C for 1 minute, 65°C for 1 minute, and 72°C for 1 minute 30 seconds followed by a final extension step of 72°C for 7 minutes, resulting in a product 173 base pairs in size. The conditions for tPA were 28 thermal cycles of 94°C for 45 seconds, 60°C for 45 seconds, and 72°C for 1 minute, followed by a final extension step of 72°C for 7 minutes, resulting in a product 234 base pairs in size. The resulting amplified DNA fragments were separated by electrophoresis through a 1.5% agarose gel, and the bands were visualised and scanned by a white/UV transilluminator (Ultra Violet Products, Cambridge, UK) and quantified by densitometry.
Assessment of cell surface exposure of negatively charged phospholipid
This was determined by analysis of annexin V binding to macrophages grown on coverslips by fluorescence microscopy. Briefly, monocytes were matured into macrophages, isolated as described above, on coverslips placed in six well cell culture dishes. The cells were treated with PM10, LPS, and staurosporine (8 μM) as a positive control for annexin V binding. After exposure to the test compounds for 4 hours, the cells were washed twice in PBS and then once in the annexin V binding buffer, 2 mM CaCl2 in HBSS. A drop of annexin V:propidium iodide solution (5 μl Alexa 488 conjugated annexin-V plus 1 μl propidium iodide (both Molecular Probes, Leiden, Netherlands) in 1 ml annexin V binding buffer) was added to the cells on the coverslip. The coverslip was then mounted and sealed onto a microscope slide to prevent drying of stained cells and the slides kept in the dark. The number of positively stained (red) cells was compared to the total number of cells, determined by propidium iodide staining, and expressed as the percentage of annexin V positive cells.
IL-8, tissue factor, and tPA assay
IL-8 release was determined by ELISA30 using an R&D Systems (Abingdon, UK) paired IL-8 antibody kit, as previously described. TF and tPA antigens were determined by ELISA (Quadratech Ltd, Epsom, UK) as per the manufacturer’s instructions.
Statistical analysis
The data are expressed as mean (SEM). Differences were evaluated using one way ANOVA followed by Tukey’s post-hoc test for multigroup comparisons.31 Statistical significance is reported when p < 0.05, p < 0.01, and p < 0.001, and indicated in figures as one, two, or three asterisks, respectively.
RESULTS
Interleukin-8 gene expression and secretion by macrophages and epithelial cells
Table 1 presents the results. There was some variation in responses between cell type and agonists. PM10 induced both gene expression and synthesis of IL-8 by macrophages and 16HBE cells comparable in extent to results with the other agonists. The response of A549 cells to PM10 was generally less marked.
IL-8 protein release and mRNA expression from macrophages, A549, and 16HBE cells treated with PM10 (50 μg/ml), TNFα (10 ng/ml), H2O2 (100 μM), and LPS (1 μg/ml) for 6 and 20 hours
Effect of PM10, oxidative stress, and LPS on procoagulant activity in culture supernatants
The supernatants from macrophages (fig 1A), A549 cells (fig 1B), and 16HBE cells (fig 1C) treated for 20 hours were examined in the pooled plasma recalcification time assay. PM10, and to a lesser extent H2O2, significantly reduced the macrophage mediated clotting time (fig 1A). Only H2O2 treatment media reduced the clotting time in the A549 experiments (fig 1B), whereas H2O2 and PM10 stimulated a clotting reaction in the 16HBE derived treatment media (fig 1C). LPS treatment supernatants had no significant effect in culture supernatants from all three cell types.

Clotting activity of supernatants of (A) macrophages, (B) A549, and (C) 16HBE cells treated with PM10 (50 μg/ml), H2O2 (100 μM), and LPS (1 μg/ml) for 20 hours. Each bar represents the mean (SEM) from at least three separate experiments. *p<0.05, **p<0.01, ***p<0.001.
Expression of negatively charged phospholipid by cultured cells
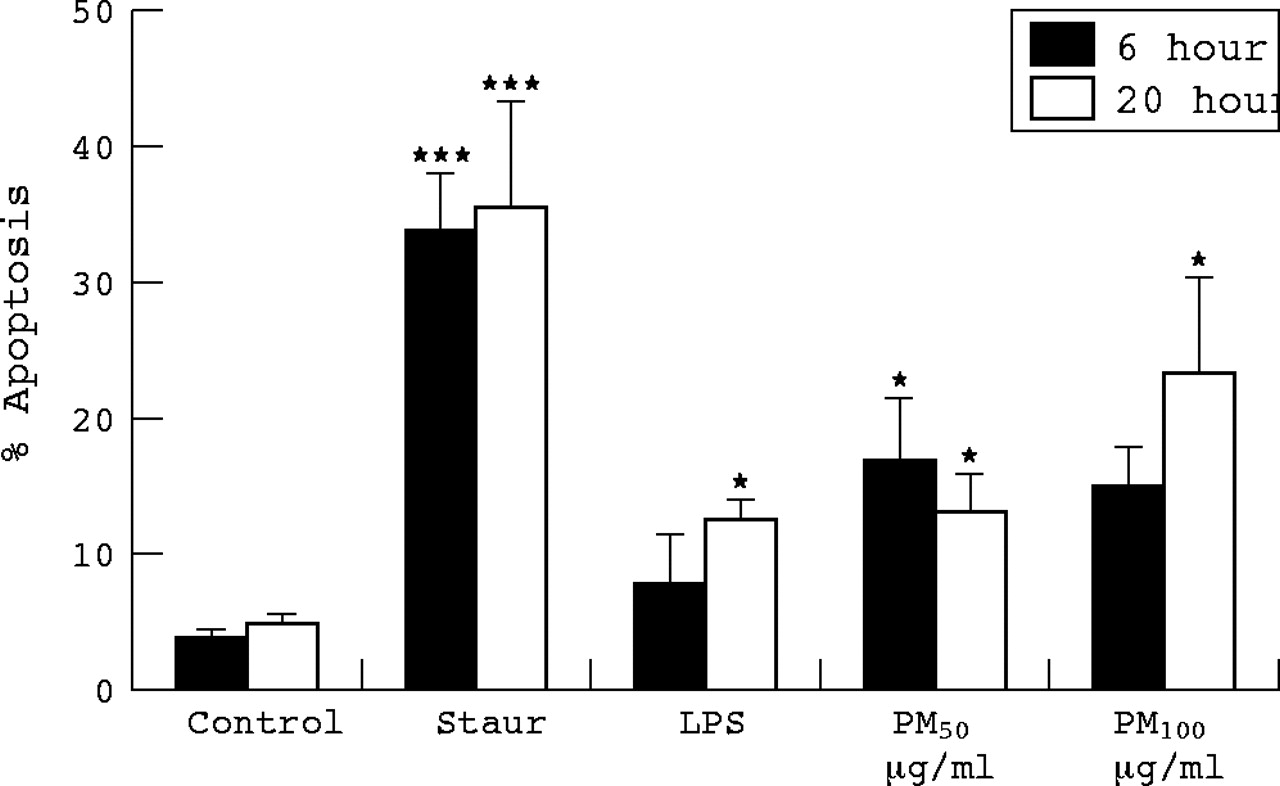
Annexin binding by macrophages after exposures for 6 and 20 hours was assessed as described in the methods (fig 2). As expected, incubation with staurosporine resulted in a marked increase in annexin V binding. Four hours treatment with PM10 at 100 μg/ml, but not 50 μg/ml, significantly increased annexin V binding. At 20 hours there was a significant increase in binding with both PM10 concentrations and at 20 hours with LPS exposure.

Percentage macrophages binding annexin V following 6 and 20 hours treatment with PM10 (50 and 100 μg/ml), H2O2 (100 μM), and LPS (1 μg/ml) as measured by Caspatag staining and counting. Each bar represents the mean (SEM) from at least three separate experiments. *p<0.05, ***p<0.001.
Macrophage tissue factor mRNA expression
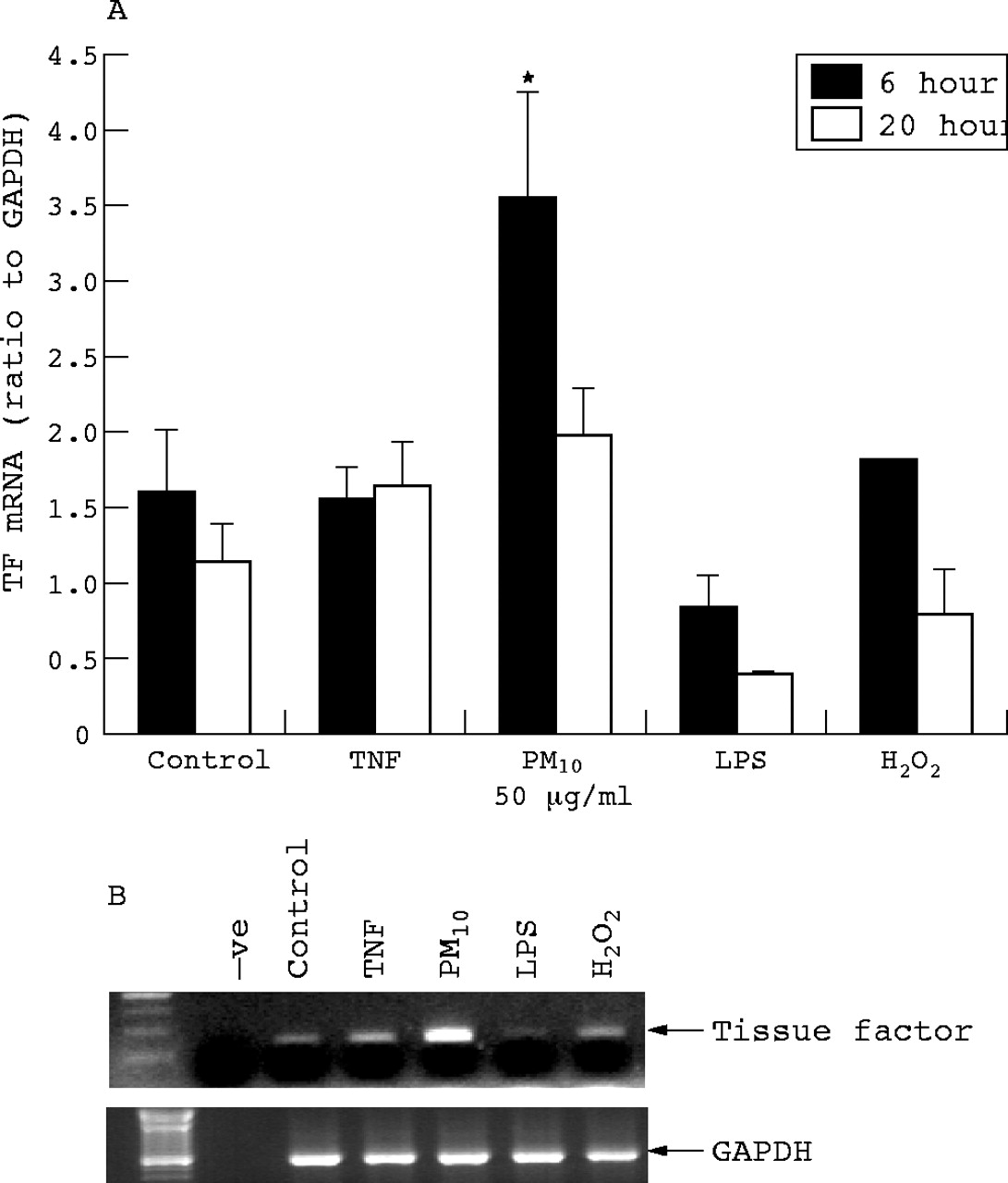
Tissue factor mRNA expression was determined in macrophages treated for 6 and 20 hours with PM10, TNF, H2O2, and LPS. At 6 hours there was a significant increase in message with PM10 only (fig 3).

Macrophage tissue factor mRNA expression as determined by RT-PCR following 6 and 20 hours treatment with PM10 (50 μg/ml), TNFα (10 ng/ml), H2O2 (100 μM), and LPS (1 μg/ml). (A) Quantification of tissue factor RT-PCR bands by densitometry. Each bar represents the mean (SEM) from at least three separate experiments. *p<0.05. (B) A representative gel from macrophages treated for 6 hours.
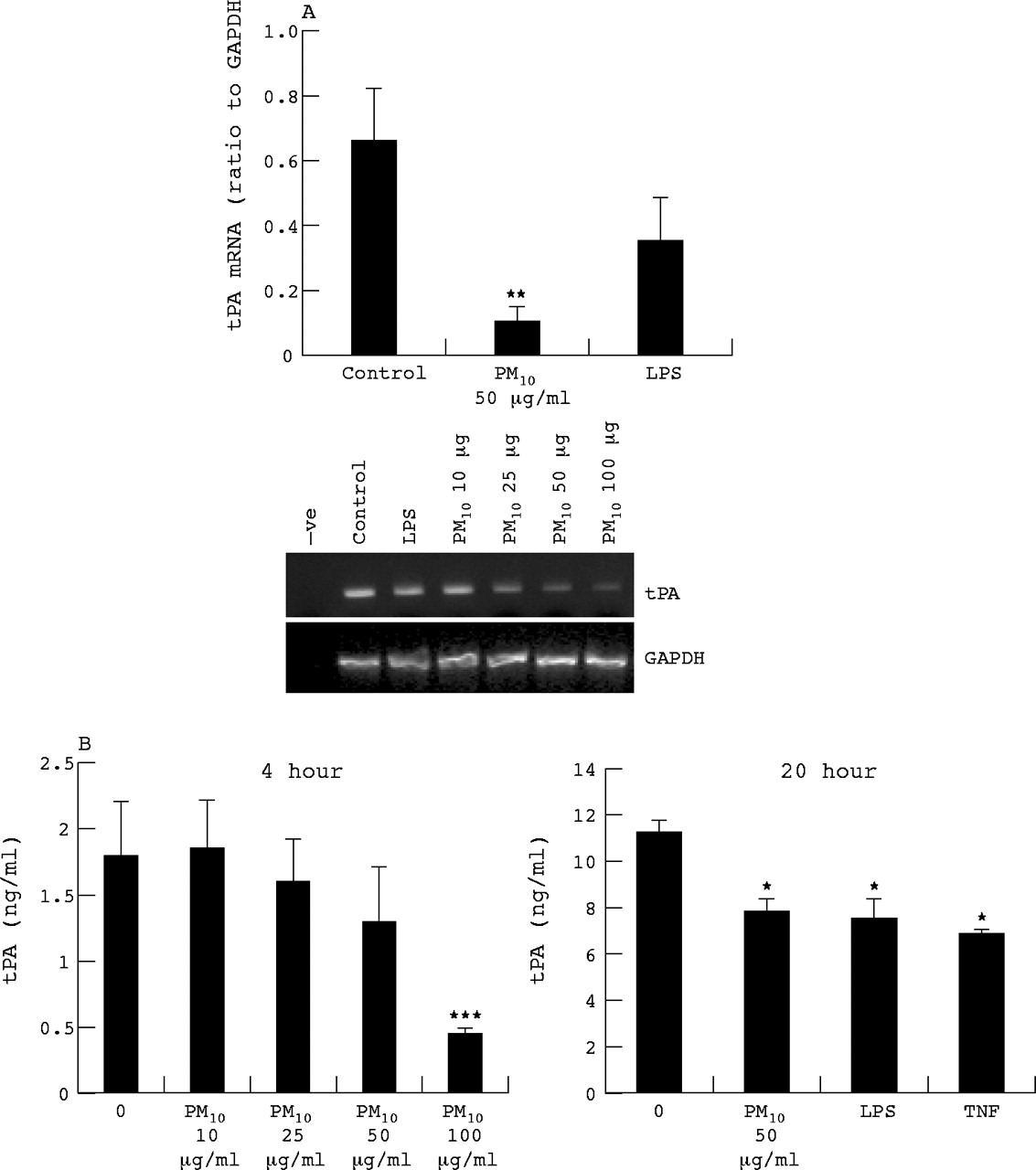
HUVEC tPA mRNA and protein expression
Vascular endothelium is the principal source of tPA. We wished to determine whether PM10 might affect vascular synthesis of tPA, should particles come into contact with endothelium within lungs, or systemically. Therefore we determined the tPA mRNA expression tPA and secretion by HUVECs treated for 6 and 20 hours with PM10 and LPS. At 6 hours PM10 reduced the expression of tPA mRNA (fig 4A) and to a greater extent than LPS treatment which also lowered tPA mRNA levels albeit not significantly. The tPA protein levels in culture medium were decreased at 4 hour PM10 treatment in a dose dependent manner, reaching significance at 100 μg/ml and also by PM10, LPS, and TNF at 20 hours (fig 4B).
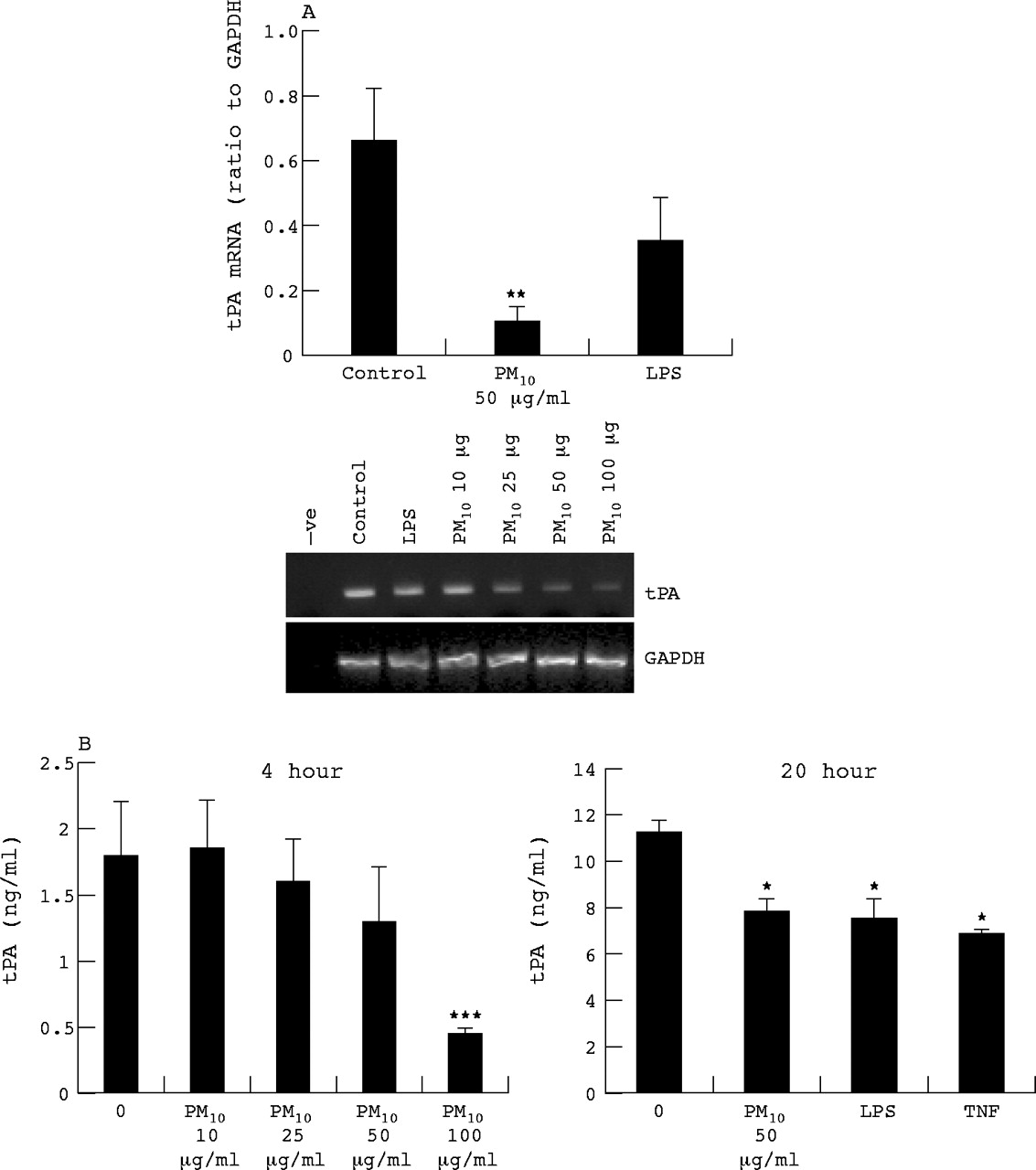
HUVEC tPA mRNA expression as determined by RT-PCR following 6 hours treatment with PM10 (10, 25, 50, and 100 μg/ml for dose response, 50 μg/ml for quantified histogram), TNFα (10 ng/ml), and LPS (1 μg/ml). (A) Quantification of tissue factor RT-PCR bands by densitometry. (B) Protein released by HUVECs as determined by ELISA following 4 or 24 hours exposure. Each bar represents the mean (SEM) from at least three separate experiments. *p<0.05, **p<0.01, ***p<0.001.
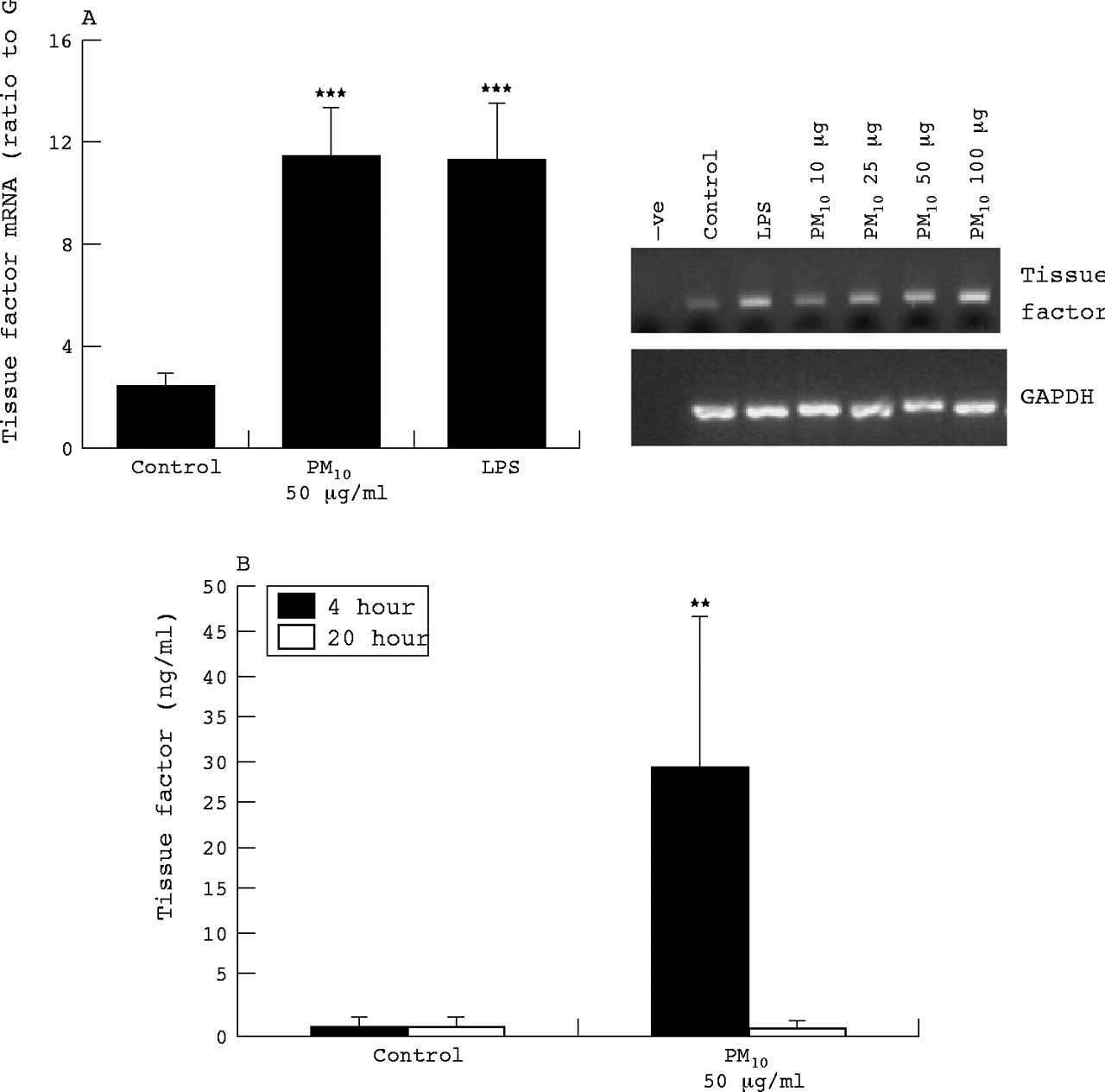
HUVEC tissue factor mRNA and protein expression
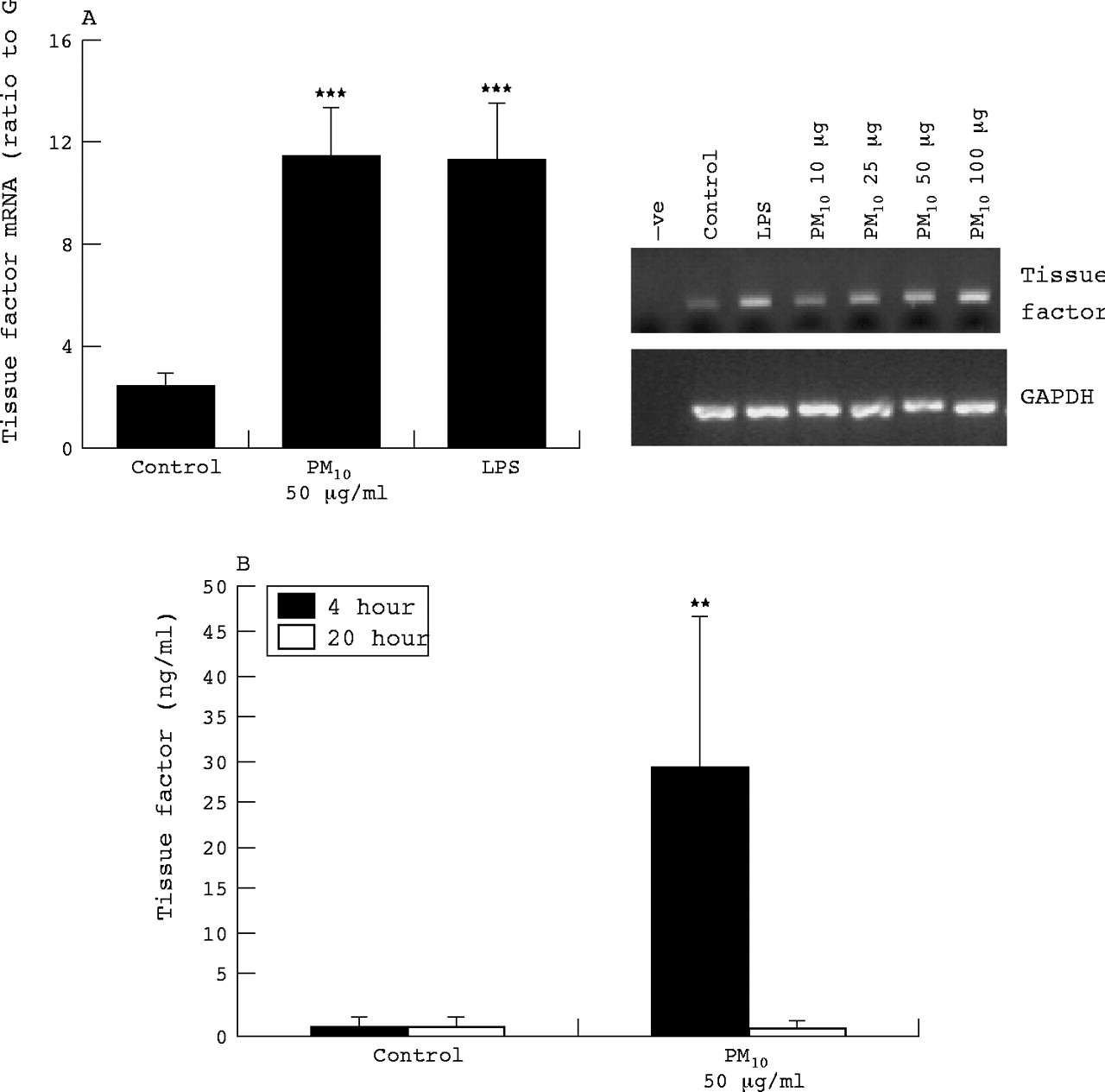
Although vascular endothelial cells do not express TF in health, there is some evidence for TF expression in vitro, in response to agonists. Therefore we also investigated TF mRNA expression and protein released into culture medium from HUVECs treated with PM10 and LPS. At 6 hours, PM10 enhanced TF mRNA expression in a dose dependent manner (fig 5A); LPS treatment also increased TF mRNA expression. The TF protein levels in culture medium at 4 hours were also increased by PM10 but were no different from control at 20 hours (fig 5B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HUVEC tissue factor mRNA expression as determined by RT-PCR following 6 hours treatment with PM10 (10, 25, 50, and 100 μg/ml for dose response, 50 μg/ml for quantified histogram), and LPS (1 μg/ml). (A) Quantification of tissue factor RT-PCR bands by densitometry. (B) Protein released by HUVECs as determined by ELISA following 4 or 24 hours exposure. Each bar represents the mean (SEM) from at least three separate experiments. **p<0.01, ***p<0.001.
DISCUSSION
Because of the link between particulate air pollution and acute cardiovascular events we wished to determine whether PM10 have potentially prothrombotic properties. First, we were able to show that supernatants from cultures of human macrophages exposed to PM10 contain procoagulant material. This property is not unique to macrophages, as a cell line of bronchial epithelial origin responded similarly. Furthermore, hydrogen peroxide exposure induced release of procoagulant material from these cells, whereas lipopolysaccharide did not. We employed a global coagulation assay in these experiments. A similar one stage clotting assay has been used previously to examine the coagulation enhancing effects of alveolar epithelial cells and macrophages from rats.32 In that study the supernatants from rat epithelial cells were found to contain more coagulation enhancing activity than isolated macrophages. However phorbol myristate acetate (PMA) was used as a stimulus and the effects of environmental particles were not considered.
In further experiments with macrophages we explored the nature of any procoagulant response to PM10. We found that these particles resulted in a significant increase in membrane surface exposure of anionic phospholipid, measured by annexin V binding. LPS was less potent, consistent with the results in the coagulation assay. In addition, tissue factor message was induced by exposure of macrophages to PM10 for 6 hours. Particle induced apoptosis in macrophages treated with environmental particles has been recently reported to be mediated by insoluble fractions of PM10.33 Although this suggests that macrophage apoptosis, mediated by contact with the particles probably through phagocytosis, may cause enhancement of coagulation, we found that staurosporine, a potent inducer of apoptosis, did not significantly increase the coagulation enhancing activity of macrophage culture supernatants (data not shown). This is consistent with the findings of Barrowcliffe and colleagues,22 who showed that although the procoagulant activity of lymphoid cell lines was related to the expression of negatively charged phospholipids, this activity was not increased when apoptosis was induced by staurosporine. Anionic phospholipid exposure is an early event in apoptosis, preceding DNA fragmentation and loss of membrane integrity,34 and our data suggest that enhanced procoagulant activity is associated with this early phase, in macrophages exposed to particles, probably through the role of anionic phospholipid in the assembly of the tenase and prothrombinase complexes.34 Our data also suggest that there may be increased tissue factor synthesis by macrophages after PM10 exposure. Furthermore, tissue factor and anionic phospholipid act synergistically in activation of coagulation as the activity of tissue factor is modulated by the surrounding phospholipid, anionic phospholipids stimulating TF-VIIa activity.35
Our results indicate that pollutant particles and other insults such as oxidative stress could generate a procoagulant environment within the lungs. It is known already that fibrin deposition, a principal end point of coagulation activation, is a characteristic of inflamed and injured lung. Furthermore, it is now recognised that there are close links between inflammation and its mediators and blood coagulation. For example, inflammatory cytokines upregulate the expression of TF, which initiates the coagulation cascade.36 It is of relevance, therefore, that we have shown that the synthesis and release of interleukin-8 from macrophages in culture is also induced by exposure to PM10. This response was also seen in the other airways cells examined, although was less marked with the alveolar cell line. The level of response was generally comparable to that seen with other agonists (LPS, TNF, and H2O2). Our finding of the induction of IL-8 production by a bronchial epithelial cell line in response to PM10 is consistent with our own recent observations and those of others that various particulate pollutants induce a proinflammatory response in lung epithelial cells.11,37–39 Interleukin-8 is a multifunctional cytokine with both proinflammatory and procoagulant properties. It is chemotactic for leucocytes and has been suggested to be an important participant in the cross-talk between the coagulation and cytokine cascades.40 It has been shown that thrombin acts as an inducer of IL-8 production in a monocyte cell line41 and also that IL-8 causes a time and dose dependent increase in procoagulant activity of monocytes.17 The generation in the pulmonary environment of inflammatory cytokines is one mechanism whereby particle inhalation could exert a systemic effect. Tissue factor also initiates an inflammatory response.42
Therefore, it seems likely that there is a synergistic role for pro-inflammatory and procoagulant factors which stimulate the activation and expression of the other. Different cell types produce different mediators to varying degrees in response to particles, and indeed are differentially exposed to particles. In this regard the combination of responses by different cell types within the complex milieu of the lung will be important in the overall resulting inflammation and procoagulant effect of particle exposure. There is precedence for a role of inflammation in enhancing a coagulation response in pneumonia patients43 and acute respiratory distress syndrome (ARDS).44
Another possible mechanism whereby inhaled particles could exert remote effects is through their passage into and distribution through the systemic circulation. It is recognised now that ultrafine particles, a component of PM10, can enter the systemic circulation in both experimental animals45 and man.46 This raises the possibility that the procoagulant effects of PM10 on macrophages might be relevant to thrombosis on atherosclerotic plaques. Macrophages are an important component of plaque, and tissue factor has been reported to be over-expressed in macrophages present in atherosclerotic plaques.47,48 Nemmar and colleagues49 have shown that blood removed from hamsters exposed to diesel particles has an enhanced ability to induce platelet aggregation.
In the light of the recent evidence that inhaled particles can enter the systemic circulation, we also postulated that particles could affect the functions of vascular endothelium. Although the concept that tissue factor can ever be synthesised by vascular endothelial cells in vivo has been challenged, in view of the likely catastrophic consequences, endothelial cells in culture do appear to be capable of expression of tissue factor when stimulated. We confirmed that LPS increases tissue factor message in cultured human umbilical vein endothelial cells. Furthermore, PM10 had a comparable effect and stimulated release of tissue factor into the culture supernatant. Perhaps of potentially greater pathophysiological significance, synthesis and secretion of the principal plasminogen activator, tPA, was inhibited in a dose dependent manner after exposure of umbilical vein endothelial cells to PM10. Whether these prothrombotic effects are present at the concentrations of particles found in vivo is open to question. There is recent evidence for particle mediated endothelial activation and for the procoagulative effects of systemic particle exposure. Particles, at a concentration relevant to the theoretical number of particles translocated to the bloodstream from a daily ambient particle exposure, introduced by intra-arterial infusion injection into the bloodstream of mice caused platelet accumulation, fibrin deposition, and fibrinogen expression in the liver.50 However, it is noteworthy that vascular endothelial cells can behave as non-professional phagocytes. This capacity could lead to concentration of particles in endothelium, as well as in macrophages in atherosclerotic plaque.
In conclusion, we have shown novel mechanisms whereby particulate air pollution may induce a procoagulant and proinflammatory response. Further studies should aim to define more precisely the relative contribution of anionic phospholipid exposure, apoptosis, and tissue factor synthesis and de-encryption, in the procoagulant activity associated with macrophage exposure to PM10.
REFERENCES
Footnotes
-
Funding: This work was supported by the British Lung Foundation, the Medical Research Council (UK), and the Colt Foundation
-
Competing interests: none declared