Article Text

Abstract
OMICS technologies are relatively new biomarker discovery tools that can be applied to study large sets of biological molecules. Their application in human observational studies (HOS) has become feasible in recent years due to a spectacular increase in the sensitivity, resolution and throughput of OMICS-based assays. Although, the number of OMICS techniques is ever expanding, the five most developed OMICS technologies are genotyping, transcriptomics, epigenomics, proteomics and metabolomics. These techniques have been applied in HOS to various extents. However, their application in occupational environmental health (OEH) research has been limited. Here, we will discuss the opportunities these new techniques provide for OEH research. In addition we will address difficulties and limitations to the interpretation of the data that is generated by OMICS technologies. To illustrate the current status of the application of OMICS in OEH research, we will provide examples of studies that used OMICS technologies to investigate human health effects of two well-known toxicants, benzene and arsenic.
Statistics from Altmetric.com
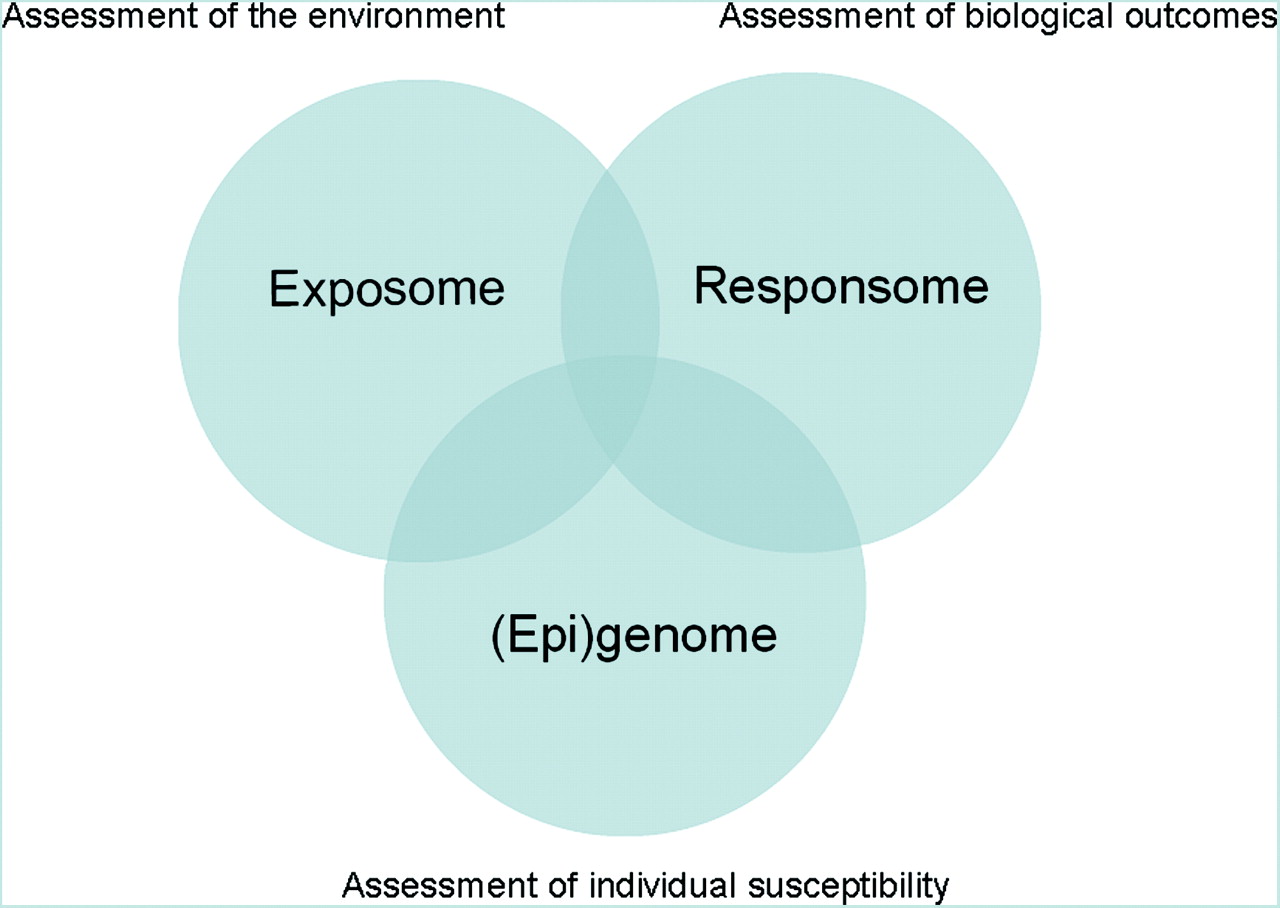
In the biological sciences the suffix -omics is used to refer to the study of large sets of biological molecules.1 The idea that the field of molecular biology needed to move from studying isolated biological molecules towards a broad analysis of large sets of biological molecules was underscored with the completion of the human genome project (HGP) in 2001.2 3 The HGP demonstrated that a relatively limited number of genes could be identified in the human genome, which substantiated the theory that complex biological processes were regulated on other levels than DNA sequence alone. This realisation triggered the rapid development of several fields in molecular biology that together are described with the term “OMICS”. The OMICS field ranges from genomics (focused on the genome) to proteomics (focused on large sets of proteins, the proteome) and metabolomics (focused on large sets of small molecules, the metabolome). We divide the field of genomics into genotyping (focused on the genome sequence), transcriptomics (focused on genomic expression) and epigenomics (focused on epigenetic regulation of genome expression). An overview of the different OMICS fields that will be discussed in this paper is presented in table 1. In this review we define the field of occupational and environmental health (OEH) research as the study of interactions between the following domains: environment (the exposome),4 individual (genetic) susceptibility (the (epi)genome), and biological outcomes (the responsome)5 (figure 1). In this context, biological outcomes can be defined as clinical diseases as well as relevant (preclinical) intermediate endpoints. In theory, OMICS technologies have a large potential value for OEH research because the environment is known to influence many of the described processes and therefore OMICS technologies are likely to provide valuable information especially where the three domains overlap. Although the field of OMICS is ever expanding (eg, see http://omics.org), currently five different OMICS fields are well established: genotyping, gene expression profiling, epigenomics, proteomics, and metabolomics. In this paper, we will address the spectacular increase in sensitivity, resolution and throughput of OMICS-based techniques in recent years, and we will discuss the difficulties regarding the interpretation of data generated by these techniques. To illustrate the current status of the application of OMICS in OEH research and the progress that has been made in recent years, we will provide examples of studies that have used OMICS technologies to investigate human health effects of two well-known environmental/occupational toxicants, benzene and arsenic.
{kind=link}
OMICS within the domain of occupational and environmental health. Genotyping operates completely within the domain of genomics. The other OMICS technologies operate in the intersection between the exposome (assessment of the environment), the responsome (assessment of health effects) and the (epi)genome (assessment of individual genetic susceptibility).
Overview of the different OMICS technologies
Overview of OMICS technologies
Genomics
We divide the field of genomics into genotyping, transcriptomics, and epigenomics.
Genotyping
Genotyping is focused on the identification of the physiological function of genes and the elucidation of the role of specific genes in disease susceptibility.6 The HGP has provided insight in the number of genes and their location in the human genome.2 3 7 This knowledge in combination with major technological improvements resulted in the development of assays that are able to assess variability in the DNA sequence of many thousands of genes in a single experiment. This development has opened the possibility to study the combined effect of variability in multiple genes on the development of complex diseases. While several types of genetic variation exist (eg, insertions and deletions of nucleotide base pairs and copy number variations (CNVs)), single nucleotide polymorphisms (SNPs) are the most commonly investigated.2 At this moment over nine million detected SNPs are available in public databases.8 9 Because SNPs are highly abundant in the human genome, they are commonly used as markers for genetic variation in disease–gene association studies.10 Due to limited genetic variation and haplotype structure and a high level of linkage disequilibrium within small regions of the genome, a subset of informative SNPs, called tag SNPs, can be genotyped as proxies for haplotype blocks to identify regional associations that influence disease or phenotypes of interest.11 Fine mapping (eg, sequencing) can further narrow the associated region in the search for the true causal variant(s). However, functional studies are needed to test whether associated SNPs alter the structure or function of DNA, RNA or proteins and influence phenotypes. Among others, functional SNPs might alter peptide sequences, transcription factor binding sites and exonic splicing enhancer/suppressor sites.
The first SNP-based studies focused on ≥1 SNPs per gene in a limited set of candidate genes. However, since the introduction of array-based genotyping techniques, allowing the simultaneous assessment of up to one million SNPs in a single assay, it has become possible to cover, with varying resolution, the entire genome in what are now commonly referred to as genome-wide association studies (GWAS). These GWAS have uncovered, and will continue to uncover, interesting and previously unknown polymorphic variants that are associated with a variety of chronic diseases. The effect sizes of these findings have in general been small (OR 1.2–1.5) fuelling debates on positive interactions between one or more common variants and the environment.12 Yet, identifying these gene–environment interactions will be difficult in ongoing GWAS given the low prevalence of exposures and/or the poor characterisation of environmental exposures in these large, often multicentre/country studies. As such, OEH research can play an important role in the identification of gene–environment interactions as the exposure is more prevalent and assessed with greater accuracy than in population- or hospital-based case-control studies that have provided most GWAS to date. Of course, sample sizes will likely be much smaller in these studies limiting the statistical power, and therefore the number of SNPs that can be tested simultaneously. Until recently most OEH studies on gene–environment have been focused on candidate genes, where the success depends on previous knowledge and ability for selection of candidate genes.13 Application of GWAS has been limited except in a study on exposure to environmental tobacco smoke.14 The application of GWAS to OEH studies will, however, result in some computational challenges as the number of genes that have a possible interaction with the exposure are large. Recently, several papers have proposed new statistical approaches for gene–environment-wide interaction studies which minimise the type 1 error (ie, false positives) while gaining efficiency and power.15–17
Although they occur less frequently than SNPs CNVs play an important role in genetic variation.18 CNVs are caused by genomic structural variations such as insertions, deletions, and duplications and have been defined as “segments of DNA that are 1 kb or larger and present at variable copy number in comparison with a reference genome”.19 CNVs located in gene promoter regions can influence gene expression, and might influence the development of complex disease traits where gene dosage is altered but not abolished.19 CNVs proximal to genes but not in promoter sequences could perturb the “histone code” and also influence gene expression. Further, CNVs located in exons could result in mis-spliced mRNA with detrimental effects on protein expression. Techniques that have been used to assess CNVs in the genome include comparative genomic hybridisation, a technique that compares labelled DNA from individuals in a study population with differently labelled reference genomic DNA,20 and SNP-based platforms that use allele intensity ratios to make inferences about CNVs.19 CNV has been frequently assessed in studies that investigated the effects of the glutathione S-transferase M1 (GSTM1) gene on environment–cancer associations.21 22 To date most studies assessed the effect of having the null genotype (deletion) of GSTM1 gene versus having at least one copy of the gene. Recent studies were also able to assess gene dosage effects (ie, does having two copies of the GSTM1 gene result in stronger associations with cancer than having one copy?).23 24
Transcriptomics
The abundance of specific mRNA transcripts in a biological sample is a reflection of the expression levels of the corresponding genes.25 Gene expression profiling is the identification and characterisation of the mixture of mRNA that is present in a specific sample. An important application of gene expression profiling is to associate differences in mRNA mixtures originating from different groups of individuals to phenotypic differences between the groups.26 In contrast to genotyping, gene expression profiling allows characterisation of the level of gene expression. Both the presence of specific forms of mRNA and the levels in which these forms occur are parameters that provide information on gene expression.27 The transcriptome in contrast to the genome is highly variable over time, between cell types and will change in response to environmental changes (table 1). A gene expression profile provides a quantitative overview of the mRNA transcripts that were present in a sample at the time of collection. Therefore, gene expression profiling can be used to determine which genes are differently expressed as a result of changes in environmental conditions. A typical gene expression profiling study includes a group of individuals with similar phenotype (eg, exposure level, disease status) and compares the gene expression profile of this group to the profile of a reference group matched on selected factors such as age and sex to the group of interest. Studies of this type usually report a set of genes that are differently expressed between the groups.
Epigenomics
The focus of epigenomics is to study epigenetic processes on a large (ultimately genome-wide) scale.28 29 Epigenetic processes are mechanisms other than changes in DNA sequence that are involved in local activity states such as gene transcription and gene silencing.30–32 Although the range of epigenetic mechanisms that are discovered is expanding, epigenomics is mainly based on two most comprehensively studied mechanisms, DNA methylation and histone modification.28 33–39 However, in recent years RNA interference of gene expression by non-coding RNAs such as microRNA and siRNA has acquired considerable attention.31 40 41 Changes in DNA methylation, histone modification and RNA interference are often associated and it is believed that interaction exists between these epigenetic processes.31 Here, the focus will be on DNA methylation and histone modification. DNA methylation is the addition of a methyl group to cytosine in a CpG dinucleotide. A distinction is made between global methylation and CpG island-specific methylation. About 70% of the CpG dinucleotides in the human genome are methylated. However, CpG dinucleotides in CpG islands are predominantly unmethylated.38 Hypermethylation of CpG islands located in promoter regions of genes is related to gene silencing. Under normal conditions gene silencing is related to phenomena such as genomic imprinting, x-chromosome inactivation and tissue-specific gene expression.28 36 Altered gene silencing plays a causal role in human disease.31 34 37 38 42 The effect of hypomethylation of the genome outside CpG islands is less well understood but may be involved in chromosomal instability.32 38 Histone proteins are involved in the structural packaging of DNA in the chromatin complex. Post-translational histone modifications such as acetylation and methylation are believed to regulate chromatin structure and therefore gene expression.34 37
Proteomics
In general the function of cells can be described by the proteins that are present in the intra- and intercellular space and the abundance of these proteins.43 Although all proteins are based on mRNA precursors, post-translational modifications (PTMs) and environmental interactions make it impossible to predict abundance of specific proteins based on gene expression analysis alone. The proteome consists of all proteins present in specific cell types or tissue. In contrast to the genome, the proteome is highly variable over time, between cell types and will change in response to changes in its environment.44 Proteomics provides insights into the role proteins have in biological systems. A major challenge is the high variability in proteins and protein abundance in certain types of biological samples (eg, the concentration of proteins in plasma ranges up to nine orders of magnitude).45 This requires the development of technologies that can detect a wide range of proteins in samples from different origins.46 Many proteomic technologies are currently available but broadly a distinction can be made between approaches that are based on detection by mass spectrometry and protein microarrays using capturing agents such as antibodies. An important focus is the identification of proteins including the presence of PTMs of proteins and identification of proteins interacting in protein complexes.43 44 Another focus of proteomics is quantification of the protein abundance. Protein expression levels represent the balance between translation and degradation of proteins in cells. It is therefore assumed that the abundance of a specific protein is related to its role in cell function. However, the high dynamic range (ie, the ratio between the smallest and largest concentration and/or mass value) of proteins complicates this type of proteomic analysis.43 44
Metabolomics
Metabolic phenotypes are the by-products that result from the interaction between genetic, environmental, lifestyle and other factors.47 The metabolome consists of small molecules (eg, lipids or vitamins) that are also known as metabolites.48 Metabolites are involved in the energy transmission in cells (metabolism) by interacting with other biological molecules following metabolic pathways. Metabolomics is defined as the study of metabolic profiles in easily collected biological samples such as urine, saliva or plasma.48 The metabolome is highly variable and time dependent, and it consists of a wide range of chemical structures (table 1). An important challenge of metabolomics is to acquire qualitative and quantitative information concerning the metabolites that occur under normal circumstances in order to be able to detect perturbations in the complement of metabolites as a result of changes in environmental factors.
Challenges for the application of OMICS in OEH
The development of new OMICS technologies is an important first step towards implementation of OMICS markers in OEH. However, similar to other (bio)markers of exposure, susceptibility and effect, the successful implementation of OMICS markers in OEH requires appropriate study designs, thorough validation of markers, and careful interpretation of study results.49–51
Study design
As indicated in table 1 the transcriptome, proteome and metabolome are highly variable over time and are likely to be influenced by the disease process. This indicates that great care should be given to the timing of biological sample collection and adequate processing (eg, field stabilisation of mRNA) of the sample to minimise measurement error and to avoid potential differential misclassification biases. In table 2 the advantages and disadvantages of the different human observational study (HOS) designs with regard to the collection and use of biological markers are given. In general, it can be stated that hospital-based case-control studies are the least suitable for the application of these technologies in HOS research, as they are more prone to selection and differential bias, while prospective studies or cross-sectional studies seem most suitable for such approaches. Moreover, hospital case-control studies are problematic as it is impossible to determine if changes in biomarkers are the cause or consequence of a disease. Semi-longitudinal studies might be extremely powerful for some OMICS technologies such as transcriptomics, proteomics and metabolomics where biological measures are taken before and after exposure or change in disease status. In these study designs each individual serves as their own control eliminating the influence of population variance.
Comparison of advantages and limitations relevant to the collection of biological specimens and data interpretation in molecular epidemiology study designs (adapted from Garcia-Closas et al, 200649)
Validation of biomarkers
The value of an OMICS-based biomarker in OEH depends on the reliability of an assay to qualitatively and quantitatively assess the biomarker and on the association between the biomarker and the biological endpoint of interest (exposure, susceptibility or health effect). The reliability of an assay can be tested by investigating the variability of an assay within and between laboratories and comparing results to the variability of existing assays (standards). A necessary step towards an increase in the reliability of OMICS assays is standardisation. Several initiatives have developed standards for new OMICS assays with regards to comparison to existing techniques (microarray quality control (MAQC)), data formats to describe experimental details (minimum information about a microarray experiment (MIAME)) and assessment of sample quality (external RNA controls consortium (ERCC)).52 53 Once the reliability of assays has been established in the laboratory transitional studies that assess the association between biomarkers and biological endpoints in humans are needed.49 To achieve an accurate estimate of the association between a biomarker and a biological endpoint reliable and valid measurements of exposure and covariates are needed as well.
A true association between a biomarker and a biological endpoint can be obscured by measurement error. To acquire insight in impact of measurement error on the observed association between a biomarker and a biological endpoint a repeated sampling design, at least on part of the population, is necessary. Repeated sampling on individuals will allow researchers to compare biomarker variability within individuals to biomarker variability between individuals. One measure that can be used to assess the variability of biomarkers within and between individuals is the intraclass correlation coefficient, which represents the proportion of the total variance that can be attributed to the between-individual variance.49 The level of measurement error that is acceptable for a biomarker depends on the magnitude of the true association between the biomarker and the biological endpoint of interest. For biomarkers with a dichotomous outcome (eg, genotyping) the accuracy of the biomarker is based on the sensitivity (eg, probability of correctly identifying an SNP) and the specificity (eg, probability of incorrectly identifying an SNP) of the biomarker.
Interpretation of study results
In recent years technological developments have had a major impact on the development of new types of study designs of OMICS-based studies. One trend that has been seen consistent within the different OMICS fields is the enormous increase in resolution of the assays (the number of “endpoints” that can be assessed in a single assay) and throughput of the assays (the number of samples that can be analysed per time period). Many of the improvements are based on the introduction of chip-based assays such as DNA microarrays. A major implication of the possibility to investigate multiple endpoints (eg, up to 1 000 000 SNPs in a single assay) in large populations is the possibility for researchers to move away from hypothesis-based studies (focused on a limited set of endpoints) towards hypothesis-free (agnostic) types of study designs (including much larger sets of endpoints). Although the hypothesis-free studies might contribute considerably to the elucidation of the complex biological processes that underlie clinically manifested health effects, it is important to realise that the interpretation of data generated by these types of studies requires a different approach than the interpretation of data generated by more traditional hypothesis-based studies. In hypothesis-based study designs “frequentist” measures such as 95% confidence intervals or p values provide a reasonably good measure to assess the statistical significance of the study's finding. However, the interpretation of such measures is based on the inclusion of a limited number of hypotheses for which the researchers assume that there is a good possibility that the null hypothesis might be rejected (ie, there is a high prior probability of a true positive finding). In a hypothesis-free analytic approach, a study is initiated without a well-defined hypothesis for each included endpoint investigated (ie, a flat prior probability for each finding). However, as a result of chance, the increased number of possible endpoints in a study is accompanied by higher probability of the possibility of detecting statistically significant false-positive results.54 Therefore, the traditional statistical approaches that are commonly used in epidemiology are of less value in hypothesis-free studies. A current challenge for the OMICS field is the development of (statistical) approaches that can be used for the interpretation of the high-dimensional data generated by these high-throughput techniques. Several statistical strategies (and also approaches in study designs) have been developed to reduce the probability of false-positive results. Examples are the Bonferroni adjustment for multiple significance testing or more sophisticated Bayesian approaches which include estimation of the false-positive report probability.15–17 54 55 However, replication of the initial findings in follow-up studies remains the strongest safeguard against false-positive results. Studies that incorporate thousands of biological endpoints should therefore primarily be seen as discovery studies that can aid the generation of new hypotheses. Therefore, new OMICS studies should incorporate strategies for built-in replication of the study findings. Application of a different analytical technique to test the hypothesis a priori in a second/validation set of samples will reduce the possibility that the initial finding was an artefact of the technology used. A potential strategy for built-in replication is to perform the initial analysis on a subset of well-characterised samples matched on potential confounders and effect modifiers and confirm the findings by using alternative analysis methods on the remaining often larger sample set. A potential problem in OEH research is, however, that replication is often complicated as there are often only a limited number of relatively small studies on a single exposure. Even if another large study can be found on a single exposure replication might still be complicated by the fact that the populations are exposed to different levels.
In addition to aspects that contribute to random error, systematic error (bias) is also a potential threat to the validity of HOS utilising OMICS technologies.56–58 The types of bias that might occur will be largely similar to types of bias that might occur in all HOS. However, issues such as sample collection, handling and storage of samples and analysis technique-specific biases might be especially relevant for studies applying OMICS technologies.57 59 60 Very recently guidelines for the reporting of genetic association studies (STREGA) have been published.61 These guidelines underline the necessity of detailed reporting in publications on genetic association studies to allow scientists to assess the potential of bias in study outcomes. Development of similar guidelines for the other OMICS fields will contribute to the identification of relevant types of bias.
Pathway analysis and systems biology
OMICS technologies will enable researchers to look at the complete complement, expression, and regulation of genes, proteins and metabolites. However, at the present time, most statistical analyses are often based on a (simplistic) one-by-one comparison of markers between exposure and/or disease groups. Recently, analytical tools/databases have become available to perform more integrated analyses of biological functions and changes in biological functions as a result of environmental factors. Examples of such approaches are gene ontology (GO), pathway analysis and structural equation modelling (SEM).62–65 GO is based on a library that consists of gene profiles that are associated with biological processes.66 Gene sets that are identified in microarray experiments as differently expressed are tested for their association with a profile in the GO library.63 In pathway analysis, not only the profile of genes associated with a specific biological process is tested, but also the functional interactions between genes in a profile.62 While still large gaps in the knowledge of biological pathways exist, each new study will contribute to build a base of knowledge necessary for these types of analyses. SEM is a statistical approach that can be used to simultaneously model multiple genes and multiple SNPs within a gene in a hierarchical manner that reflects their underlying role in a biological system.65
The increasing knowledge of biological pathways will facilitate the integration of the separate OMICS fields into systems biology approaches. System biology has been described as a global quantitative analysis of the interaction of all components in a biological system to determine its phenotype.67–69 This integration is facilitated by a continuous increase in computing power and possibilities for data sharing.
Examples of the use of OMICS in occupational and environmental health research
In table 3 a number of studies are listed to illustrate the current application of OMICS technologies in OEH research. Benzene and arsenic were chosen as examples because of the large populations with potential exposure to these agents in both the occupational and environmental setting and the relatively large number of studies on these agents that have applied OMICS technologies. It should be noted that inclusion of the example studies was not intended as a systematic overview of studies applying OMICS in OEH research in these specific areas but merely to provide a resource of studies that are indicative of the potential of these new technologies. We highlight three studies from table 3 in some more detail to illustrate the progress in the OMICS field that has been made in recent years. A nice illustration of the progress of the use of genotyping methods in OEH research is a study on haematological effect among a cohort of 250 workers exposed to benzene and 140 controls.70–72 Initial gene–environment analyses in this study were based on candidate gene approaches focusing on genes involved in the metabolism of benzene (four genes, four SNPs),72 DNA double strand break repair (seven genes, 24 SNPs),71 and cytokine and cellular adhesion molecule pathways (20 genes, 40 SNPs).70 In a more recent analysis of the same study population, Lan et al used a chip-based assay (GoldenGate assay) for genotyping which allowed for a larger number of SNPs to be assessed (414 genes, 1433 SNPs).73 These SNPs were selected from the SNP500Cancer database, and were, therefore, hypothesised to be involved in the development of cancer. However, the influence of these SNPs on benzene-induced haematotoxicity was largely unknown for most SNPs. This study should therefore primarily be seen as hypothesis generating and indeed has provided information on several putative genes involved in benzene haematotoxicity that went well beyond the more classical focus in OEH research on metabolic genes. Although the authors addressed issues of multiple comparisons to reduce the chance of false-positive findings due to the large number of SNPs included in the analysis, it is still critical that the results are replicated in subsequent independent studies.
Examples of the use of OMICS technologies in occupational and environmental studies that investigate health effects in human populations exposed to benzene or arsenic
An example of a hypothesis-free approach towards the assessment of the transcriptome comes from a study by Argos et al.74 In this micro-array-based study ∼22 000 genome-wide gene transcripts were measured in 25 subjects with arsenic-induced skin lesions and 15 controls. A false discovery rate of 1% was defined a priori to reduce the risk of chance findings. A set of 486 genes that were differentially expressed between cases and controls was reported. The gene transcripts were also analysed with the use of gene ontology and pathway analysis approaches to elucidate the biological pathways that are involved in arsenic-induced skin lesions. Similar to the genotyping results of the studies discussed above, results from the genome-wide assessment of the transcriptome should be interpreted with great care and require replication in independent studies before they can be used as valid exposure or effect markers.75 76
Way forward
It is clear that there have been great technological advances in the different OMICS fields. Some of these technologies have and are starting to be applied in OEH research and will undoubtedly lead to numerous new insights in the near future. With the development of validated technologies, appropriate study designs, better sample handling and advanced statistical methods for data interpretation, OMICS techniques will eventually contribute significantly to OEH and will help the field progress towards an integrated view of the interaction between environment and human health. To achieve this integrated view it will be important to not only focus on genetic variants but also on more functional measures of the phenotype and accurate assessment of exposure. The challenge in this effort will be that the closer one gets to a functional measure of the phenotype (ie, proteomics, metabolomics) the more complex it will be to capture physiologically relevant variability and the more crucial the development of advanced study designs, sampling collection procedures, measurement techniques, and methods for statistical analysis will be to allow interpretation of these parameters.
Acknowledgments
This work was performed as part of the work package “integrated risk assessment” of the ECNIS Network of Excellence (Environmental Cancer Risk, Nutrition and Individual Susceptibility), operating within the European Union 6th Framework Program, Priority 5: “Food Quality and Safety” (FOOD-CT-2005-513943).
References
Footnotes
Funding European Union 6th Framework Program “ECNIS” (FOOD-CT-2005-513943). MTS, LZ and CFS were supported by NIH grants P42ES004705, R01 ES006721, R01 CA122663, and U54 ES016115.
Competing interests MTS has received consulting and expert testimony fees from law firms representing both plaintiffs and defendants in cases involving exposure to benzene.
Provenance and peer review Not commissioned; externally peer reviewed.